Autocoids -- Histamine
Histamine Pharmacology
1. Introduction to Autacoids
What is an Autacoid?
The term comes from the Greek words Autos (meaning "self") and Akos (meaning "medicinal agent" or "remedy"). Therefore, an autacoid is literally a "self-remedy."
By definition, Autacoids are endogenous substances (made naturally inside the body) that act as biological factors or "local hormones".
A classic hormone (like insulin or thyroid hormone) is produced in a specific, centralized gland, dumped into the systemic bloodstream, and travels a long distance to reach its target organ.
Autacoids are DIFFERENT:
- They are produced by widely distributed tissues all over the body, not a single gland.
- They act locally (at or very close to their exact site of synthesis and release).
- They are present in very small amounts.
- They have a short lifespan with a very short duration of action (they are rapidly destroyed to prevent them from causing systemic chaos).
Note: However, if produced in massive, pathological amounts (like during severe anaphylactic shock), they can overcome local destruction, enter the systemic circulation, and have life-threatening systemic effects.
Classification & Examples of Autacoids
You must know the chemical classification of the different autacoids. Exam questions frequently mix these up:
| Chemical Class | Examples |
|---|---|
| Amines | Histamine, Serotonin (5-HT) |
| Polypeptides (Proteins) | Kinins (Bradykinin), Oxytocin, Angiotensin, Vasopressin, Endothelins |
| Fatty Acids (Eicosanoids) | Prostaglandins, Leukotrienes, Thromboxanes, Platelet Activating Factor (PAF) |
| Others | Nitric Oxide (NO - Endothelium-derived relaxing factor), Cytokines |
2. Histamine: Synthesis, Storage, and Metabolism
Histamine is a ubiquitous molecule. It is present everywhere: in bacteria, plants, animals, and notably in venoms and stinging fluids (like bee stings, wasp venom, or stinging nettle plants).
Chemistry & Synthesis
- Chemistry: It is a basic amine, specifically a β-aminoethylimidazole.
- Synthesis: The amino acid L-Histidine undergoes decarboxylation (the chemical removal of a CO2 molecule) to become Histamine. The specific enzyme that performs this action is L-Histidine decarboxylase.
Inactivation & Metabolism
Because histamine is so incredibly potent, it must be deactivated rapidly if it isn't safely stored away. There are two major metabolic pathways the body uses to break it down and excrete it in the urine:
- Pathway 1 (Methylation): Conversion to N-methylhistamine (via the enzyme N-methyl transferase), which is then oxidized by MAO (Monoamine Oxidase) / DAO into methylimidazoleacetic acid.
- Pathway 2 (Oxidation): Direct conversion by the enzyme Diamine Oxidase (DAO) into imidazoleacetic acid (IAA).
3. Histamine Storage and Release Mechanisms
Where is histamine kept? In humans, it is mostly stored inside Mast Cells (found abundantly in tissues interfacing with the outside world like Skin, Lungs, and GI tract) and Basophils (circulating in the blood). Inside these cells, histamine is locked up in granules, tightly bound to a heparin-protein complex so it doesn't leak out.
Histamine can be released in two distinct ways: Immunologic (Antigen-mediated) and Non-Immunologic.
A. Immunologic Release (Antigen-Mediated)
This is the classic Type I Hypersensitivity (Immediate Allergic Reaction).
- The Process: A person is exposed to an allergen (e.g., pollen, peanuts). Their immune system mistakenly creates IgE antibodies against it. These IgE antibodies attach to the surface of mast cells (a process called sensitizing the cell). Upon a second exposure to the same pollen, the allergen physically bridges and cross-links the IgE antibodies on the mast cell surface.
- The Result: The mast cell degranulates "explosively", dumping massive amounts of histamine into the tissue. This specific process is energy-dependent (requires ATP) and requires calcium.
Negative Feedback & The Lung Exception
In skin mast cells and blood basophils, the released histamine eventually binds back onto its own H2 receptors located on the mast cell's own surface. This acts as a biological "brakes" system, inhibiting further histamine release (Negative Feedback).
EXAM EXCEPTION: This feedback inhibition does NOT occur in lung mast cells! This is exactly why allergic asthma attacks in the lungs can spiral out of control so rapidly and become fatal; there are no built-in brakes to stop the continuous histamine release in the bronchioles.
B. Non-Antigen Mediated Release
This release mechanism does not require the immune system to be sensitized with IgE. It happens through direct physical or chemical interaction.
- Chemical Release: Certain drugs and chemicals can physically enter the mast cell and displace histamine from its heparin complex, forcing it out.
- Examples: Morphine, Tubocurarine (neuromuscular blocker), radiocontrast media (used in CT scans), amides, alkaloids, and basic polypeptides (like wasp/bee venoms).
- Mechanical Release: Physical trauma forces the mast cells to burst open. Examples: Vigorous scratching of the skin, severe burns, or crushing injuries.
- Cellular Proliferation: Pathological overgrowth of cells naturally increases total body histamine levels simply because there are more cells making it. Examples: Leukemia, Gastric Carcinoid Tumors.
- Physical Stimuli: Extreme cold, excessive heat, or exposure to bacterial toxins.
"Red Man Syndrome" & IV Morphine
The Event: If a nurse pushes an intravenous dose of Morphine too fast, the patient may suddenly flush bright red, feel intensely hot, become incredibly itchy, and their blood pressure might drop precipitously.
The Mechanism: This is frequently mistaken for an allergy. It is not a true allergy (no IgE is involved). The rapid bolus of morphine chemically displaced histamine from the patient's mast cells all at once, causing sudden, massive vasodilation. This is a classic example of Non-Antigen Mediated Chemical Release.
The Fix: Stop the infusion, administer an antihistamine (like Diphenhydramine), and when restarting, push the morphine much slower.
4. Sites of Histamine Action
Histamine regulates multiple physiological systems beyond just making you sneeze:
- Mast Cells & Basophils: Triggers standard inflammation and allergy symptoms (Skin itching, Lung wheezing, GIT cramping).
- Central Nervous System (CNS): Acts as a critical neurotransmitter, keeping the brain awake and alert.
- Neuroendocrine: Regulates hormones. It stimulates the release of ACTH, Prolactin (PRL), Vasopressin (VP), Oxytocin, and LH. It inhibits the release of GH and TSH.
- Thermal & Cardio: Causes hyperthermia (feverish feeling) via H1/H3 receptors located in the preoptic nucleus of the hypothalamus.
- Body Weight & Sleep: Acts as a powerful appetite suppressant (via H1), potentiates the hormone leptin (causing weight loss signaling), accelerates lipolysis (fat breakdown), and regulates sleep/arousal (keeps you awake).
- Stomach: Released from entero-chromaffin-like (ECL) cells in the stomach wall. It is one of the primary secretagogues that activate parietal cells to pump out massive amounts of gastric acid.
5. Histamine Receptors & Their Effects
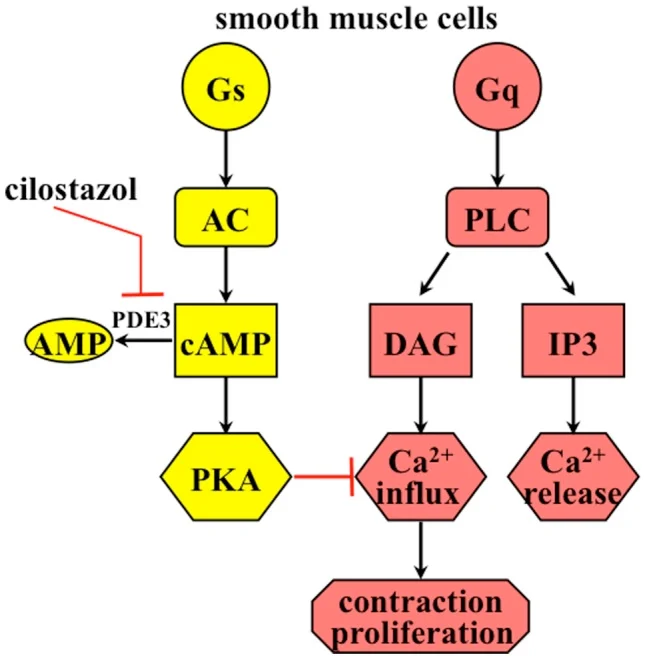
Histamine acts on four distinct receptors (H1, H2, H3, H4). ALL of them are G-Protein Coupled Receptors (GPCRs). Currently, clinical pharmacology heavily targets H1 and H2.
| Receptor | Location / Distribution | Post-Receptor Mechanism | Selective Antagonists (Blockers) |
|---|---|---|---|
| H1 | Smooth muscle (bronchi, gut), Endothelium, Brain | Gq → ↑ IP3, DAG → ↑ Intracellular Ca2+ | Mepyramine, Cetirizine, Loratadine |
| H2 | Gastric mucosa (parietal cells), Cardiac muscle, Mast cells, Brain | Gs → ↑ cAMP | Ranitidine, Cimetidine, Famotidine |
| H3 | Presynaptic neurons (Brain, myenteric plexus) | Gi → ↓ cAMP, ↓ Ca2+ | Thioperamide |
A. H1-Receptor Stimulation (The Allergy Receptor)
When histamine hits H1 receptors, it causes severe, rapid inflammatory changes:
- Endothelial Contraction: The endothelial cells lining venules actually shrink and pull apart, widening the gaps between them. This drastically increases vascular permeability, allowing protein-rich fluid to leak out into the tissues (this causes edema/swelling and a runny nose).
- Smooth Muscle Contraction: Causes severe bronchoconstriction (asthma attack), intestinal cramps (diarrhea), and uterine contractions.
- Vasodilation: Despite contracting the venules, it heavily dilates the arterioles. This causes the classic red flushing, severe headaches (vessels in the brain swelling), and a dangerous drop in blood pressure.
- Nerve Endings: Stimulates superficial sensory nerves to cause Pain and intense Itching (Pruritus).
The Triple Response of Lewis
If you take a dull instrument and firmly scratch a person's skin, histamine is released locally. This causes three distinct, highly predictable visual phases to appear on the skin:
- Flush (Red Spot): A localized red spot appears instantly along the scratch line due to direct capillary vasodilation.
- Weal (Swelling/Bump): The scratched area raises up and becomes puffy due to vascular leakage (edema) caused by endothelial contraction.
- Flare (Red Halo): A much wider, brighter red area spreads outwards surrounding the scratch. This is caused by indirect vasodilation (an axon reflex triggering nearby vessels to also dilate).
B. H2-Receptor Stimulation (The Stomach Receptor)
- Stomach: Activates Parietal Cells to massively secrete H+ (stomach acid). This is the major target for ulcer-healing drugs.
- Heart: Increases the force of contraction (positive inotropy) and increases Heart Rate (positive chronotropy).
- Blood Vessels: Causes vasodilation.
C. H3-Receptor Stimulation (The Brain/Nerve Receptor)
H3 receptors are mostly presynaptic (they sit on the nerve terminal that is releasing the chemical, acting as volume control knobs).
- Autoreceptors: When histamine binds to an H3 autoreceptor on a histamine-releasing neuron, it provides negative feedback, stopping the synthesis and release of more histamine.
- Heteroreceptors: When histamine binds to H3 receptors on *other* nerve types, it inhibits the release of other major neurotransmitters like GABA, Norepinephrine, Dopamine, Serotonin, and Acetylcholine.
Future Pharmacology: H3 Agonists
Because H3 receptors regulate brain chemistry so heavily, they are massive potential therapeutic targets for cognitive and psychiatric disorders such as Sleep disorders (Narcolepsy), Parkinson's disease, ADHD, and Schizophrenia.
Examples of H3 Agonists:
- α-methylhistamine
- Cipralisant
- Imbutamine (also an H4 agonist)
- Immepip
- Imetit
- Immethridine
- Methimepip
- Proxyfan
6. Pathological Reactions & Clinical Uses of Histamine
Pathology Mediated by Histamine
- Type I Hypersensitivity: Hay fever, allergic rhinitis (itchy/watery eyes, sneezing), urticaria (hives from nettles or insect stings).
- Anaphylactic Shock: Massive systemic histamine release causing severe hypotension (shock from vasodilation) and suffocation (from severe bronchoconstriction).
- Emesis: Histamine mediates motion sickness pathways in the brain.
- Peptic Ulcer Disease (PUD): Excessive H2 stimulation causes an acid overload, eating through the protective stomach lining.
Clinical Uses of Pure Histamine
Doctors rarely give pure histamine as a treatment because it is highly uncomfortable and dangerous (it causes shock and asthma). However, it has one specific diagnostic use:
Diagnostic Positive Control: It is used as a positive control injection during allergy skin testing. If a doctor is trying to see what you are allergic to, they will prick your back with 20 different allergens. They will also prick you with pure histamine. If the pure histamine prick doesn't produce a Weal and Flare, it means either your immune system is completely unresponsive, or you cheated and took an antihistamine pill before the test, rendering the entire allergy test invalid.
7. Antagonists (The "Antihistamines")
A. H1 Antagonists (Allergy & Cold Meds)
These drugs competitively block histamine from binding to H1 receptors. They reliably relieve sneezing, itchy eyes, runny nose, and hives. They are also used for allergies, motion sickness, vertigo, and insomnia.
They are divided into two distinct generations based heavily on their ability to cross the Blood-Brain Barrier (BBB).
The Sedating Ones
These are lipophilic, cross the BBB easily, block H1 in the brain (causing profound sleepiness), and often lack specificity (they also block muscarinic receptors, causing dry mouth, blurred vision, and urinary retention).
- Highly Sedative & Potent: Promethazine, Hydroxyzine, Diphenhydramine, Dimenhydrinate (great for motion sickness).
- Moderately Sedative: Pheniramine, Cinnarizine, Meclizine, Buclizine, Cyproheptadine (unique because it also stimulates appetite).
- Mild/Less Sedative: Chlorpheniramine, Dexchlorpheniramine, Clemastine, Mebhydroline, Dimethindone.
The Non-Sedating Ones
These are bulky or ionized molecules that do not cross the BBB well. They are mainly pure anti-allergics with little to no sleepiness and fewer muscarinic side effects.
- Examples: Cetirizine, Levocetirizine, Loratadine, Desloratadine, Fexofenadine, Azelastine, Ebastine, Mizolastine, Rupatadine.
Clinical Application of H1 Blockers
The Truck Driver: If a commercial truck driver has bad seasonal allergies, you MUST NOT prescribe Diphenhydramine (1st gen), or he will fall asleep at the wheel and crash. You must prescribe Loratadine or Fexofenadine (2nd gen).
The Itchy Sleepless Patient: Conversely, if a patient cannot sleep because they are covered in an incredibly itchy poison ivy rash, Diphenhydramine is the absolutely perfect drug because it cures the itch *and* utilizes its sedative side effect to help them sleep.
For Vertigo/Migraines: Flunarizine and Cinnarizine are specifically noted for having excellent antivertigo and antimigraine properties by regulating inner ear fluid and blood flow.
B. H2 Antagonists (The Acid Blockers)
H2 blockers profoundly reduce stomach acid production by competitively blocking histamine at the H2 receptors on the stomach's parietal lining. They are primarily used to treat heartburn, Gastroesophageal Reflux Disease (GERD), peptic ulcers, and indigestion.
Parietal Cell Mechanism (Why H2 blockers work so well)
- ACh & Gastrin → bind to receptors → increase Intracellular Calcium (Ca2+)
- Histamine → binds H2 Receptor → increases cAMP (via ATP)
- Convergence: Both of these pathways ultimately converge to turn ON the Gastric K+/H+ Ion Pump (the Proton Pump), actively dumping severe acid (H+) into the stomach.
- By taking an H2 blocker, you sever the cAMP pathway, heavily crippling the parietal cell's ability to produce acid, allowing the ulcer to heal.
The "Tidine" Family (Table 62-1 Comparison)
You must know the relative potencies and dosing strategies of these drugs:
| Drug | Relative Potency | Typical Acute Ulcer Dose | GERD Dose |
|---|---|---|---|
| Cimetidine | 1 (Least Potent) | 800 mg HS (at bedtime) or 400 mg bid (twice daily) | 800 mg bid |
| Ranitidine | 4 - 10x stronger | 300 mg HS or 150 mg bid | 150 mg bid |
| Nizatidine | 4 - 10x stronger | 300 mg HS or 150 mg bid | 150 mg bid |
| Famotidine | 20 - 50x stronger (Most Potent) | 40 mg HS or 20 mg bid | 20 mg bid |
Cimetidine Side Effects
Although it is the historical prototype H2 blocker, Cimetidine is famous on pharmacology exams primarily for its negative side effects.
- It heavily inhibits Cytochrome P450 enzymes in the liver, causing massive drug interactions by preventing the breakdown of other drugs (like Warfarin or Diazepam), leading to toxicity.
- It has strong anti-androgenic effects (it blocks testosterone receptors). In men, chronic use can cause gynecomastia (breast tissue growth), decreased libido, and impotence.
Because of these issues, Ranitidine or Famotidine are usually preferred clinically, as they lack these severe side effects while being much more potent.
Quick Quiz
Histamine Quiz
Pharmacology - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Histamine Quiz
Pharmacology
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.