The Perineum
Complete, exhaustive anatomical study covering boundaries, fascia, urogenital and anal triangles, neurovascular supply, and clinical applications.
SECTION 01: Boundaries & Surface Anatomy
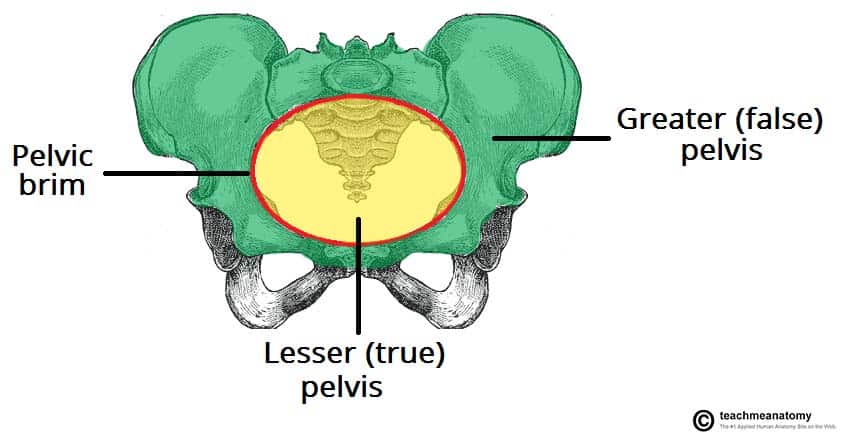
The perineum is the diamond-shaped region located inferior to the pelvic diaphragm, representing the lowest partition of the trunk. It is bounded by the pelvic outlet and is separated into two distinct triangular sub-regions by a theoretical transverse line connecting the ischial tuberosities.
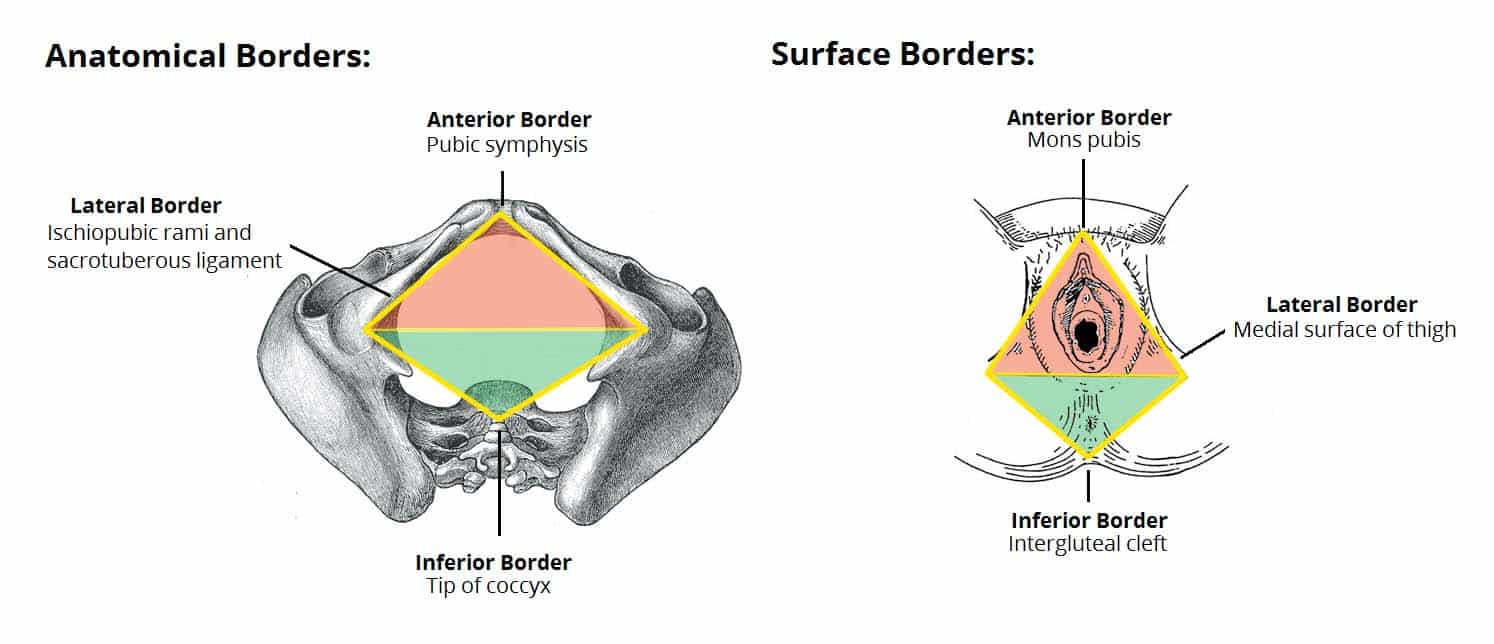
Perineal Boundaries
The perineum is defined by the following osseofibrous borders:
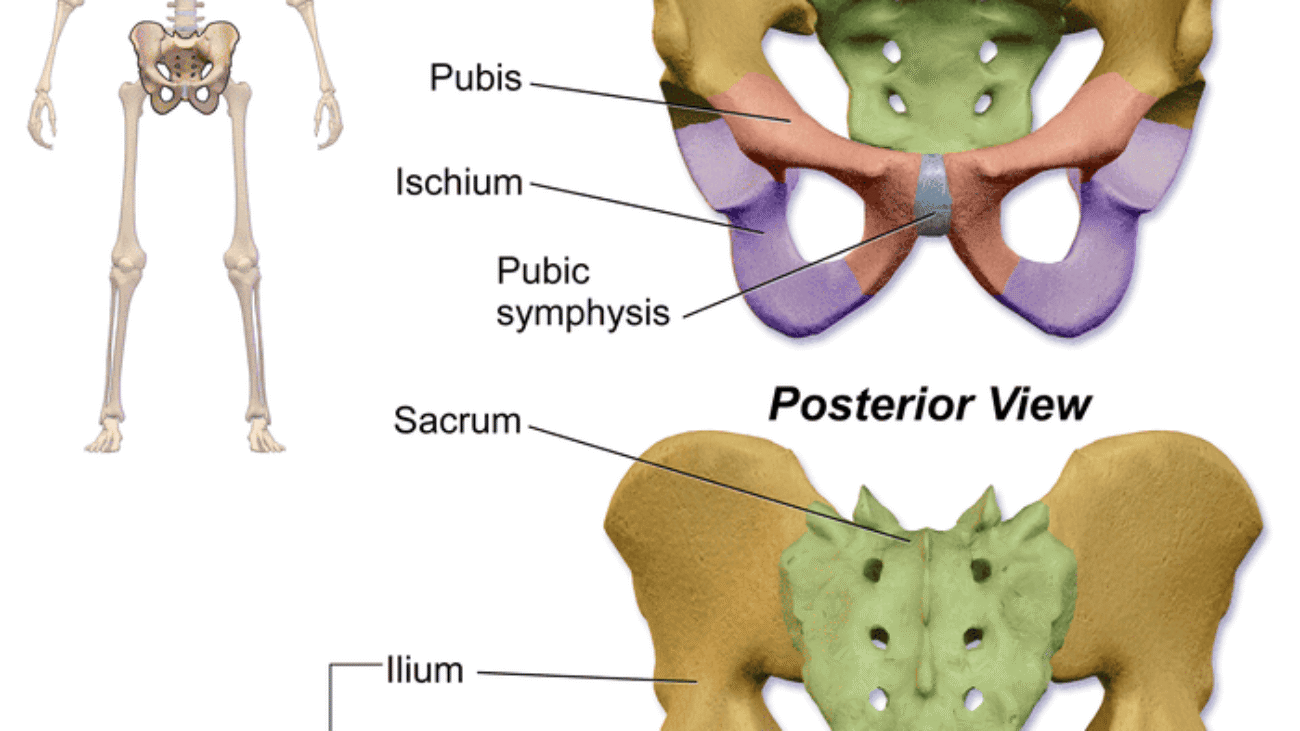
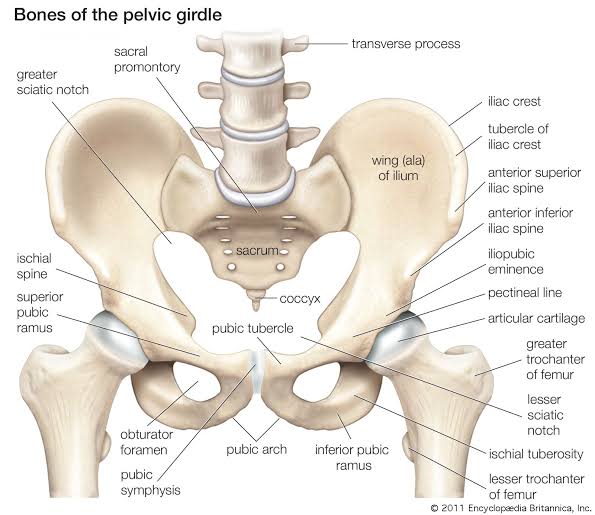
- Anterior Boundary: Pubic symphysis — The secondary cartilaginous joint between the left and right pubic bones. The perineum begins immediately posterior to this structure.
- Anterolateral Boundaries: Inferior pubic rami and ischial rami (ischiopubic rami) — The fused inferior pubic and ischial rami form the bony sides of the anterior perineum.
- Lateral Boundaries: Ischial tuberosities — The weight-bearing bony prominences that serve as the lateral corners of the perineal diamond. These are the key landmarks for dividing the perineum into triangles.
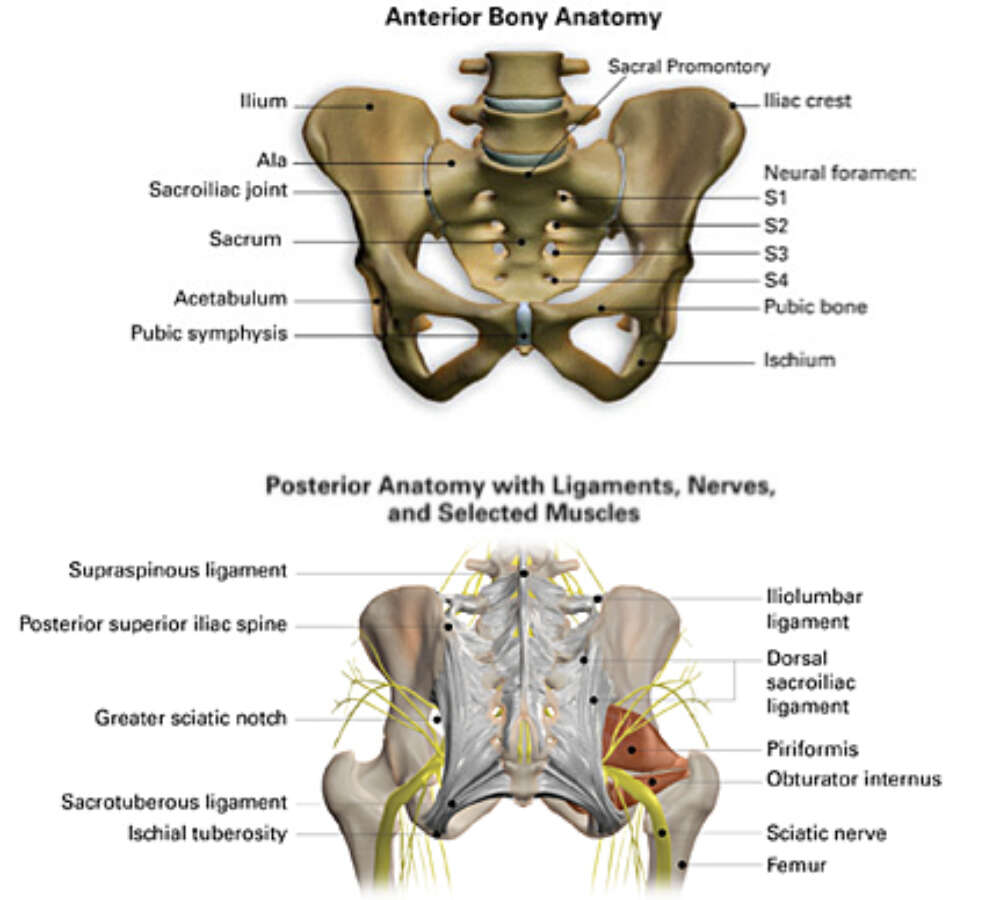
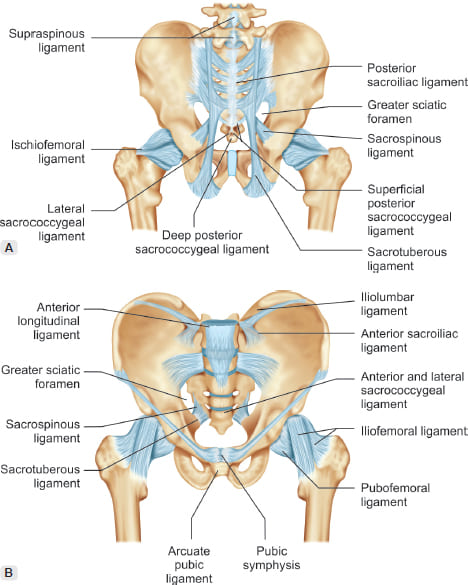
- Posterolateral Boundaries: Sacrotuberous ligaments — The strong fibrous bands extending from the sacrum to the ischial tuberosities, forming the posterolateral margins.
- Posterior Boundary: Apex of the coccyx — The terminal tip of the vertebral column, forming the posterior apex of the perineal diamond.
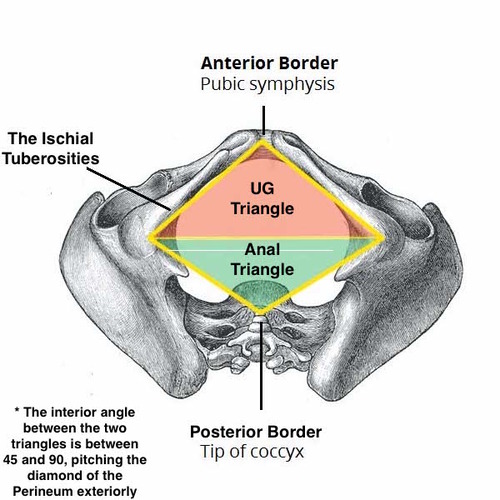
Divisions of the Perineum
An imaginary transverse line connecting the two ischial tuberosities divides the diamond-shaped perineum into two triangles:
Urogenital Triangle (Anterior)
- Directed downward and forward.
- Contains the external genitalia and urethral opening.
- Bounded by pubic symphysis and ischiopubic rami.
- Base is the line between ischial tuberosities.
- Apex is the pubic symphysis.
- Further divided into superficial and deep perineal spaces.
Anal Triangle (Posterior)
- Directed downward and backward.
- Contains the anal canal and its opening (anus).
- Bounded by sacrotuberous ligaments and coccyx.
- Base is the line between ischial tuberosities.
- Apex is the coccyx.
- Contains the ischioanal fossae on either side of the anal canal.
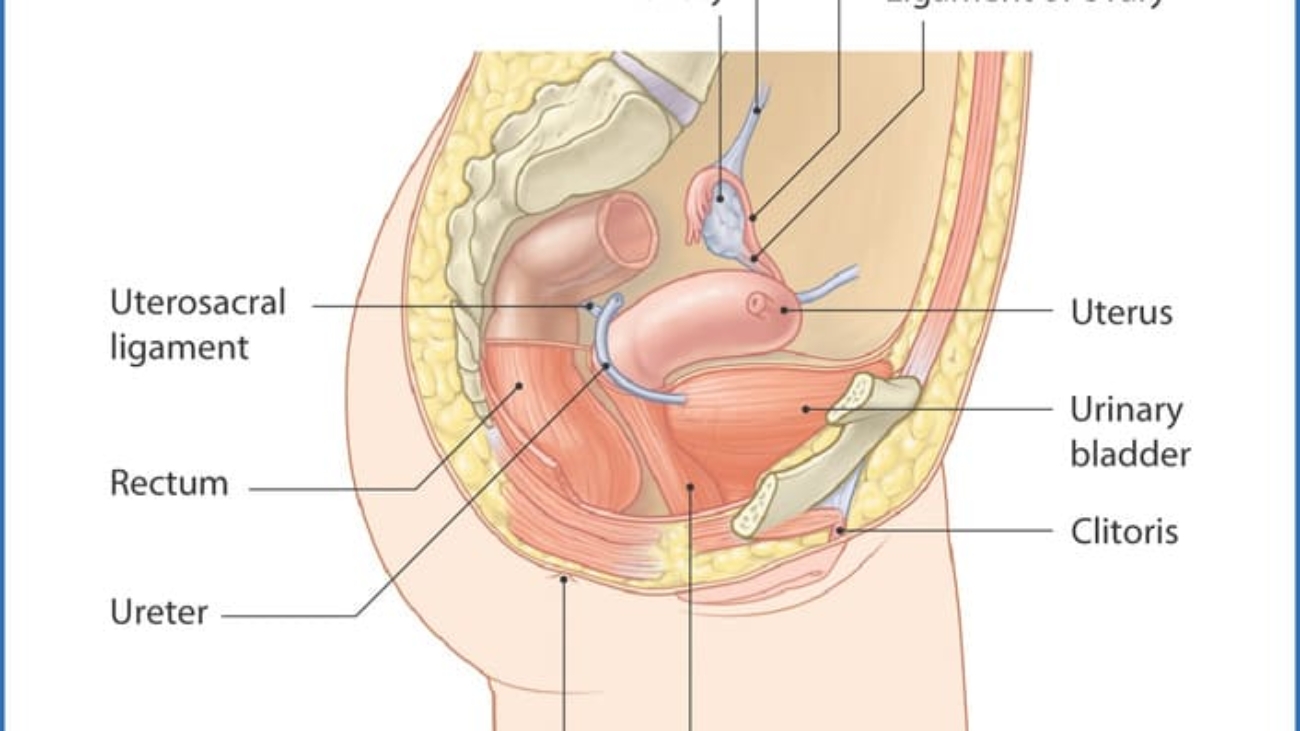
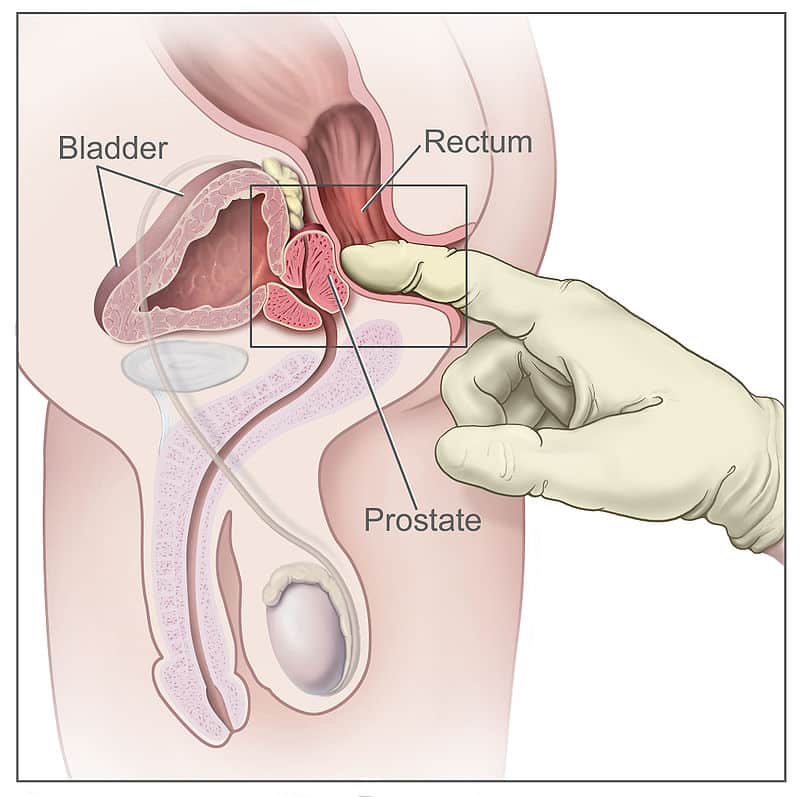
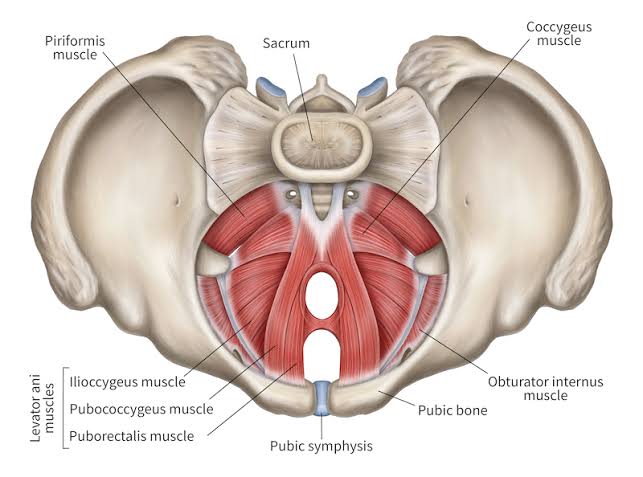
Perineal Body (Central Tendon of Perineum)
A fibromuscular mass located in the midline at the junction between the urogenital and anal triangles. It is the central anchoring point of the perineum and serves as the attachment site for multiple muscles. It is approximately 2-3 cm in diameter and lies about 2 cm anterior to the anus in females.
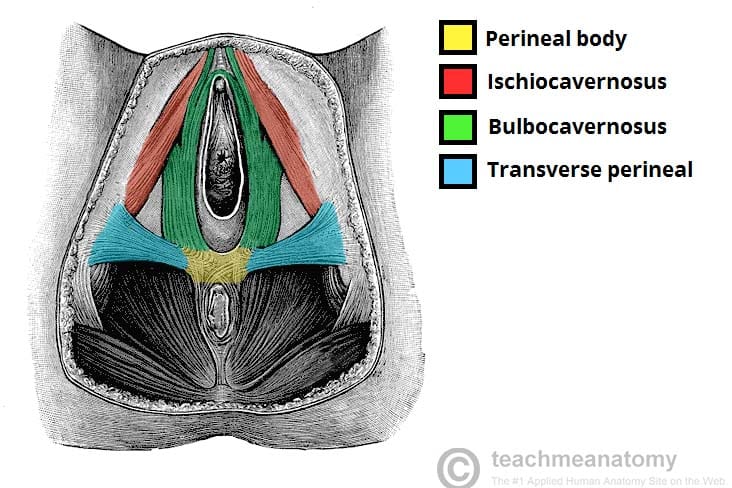
Muscles that attach to or anchor into the perineal body:
| Muscle | Origin/Insertion at Perineal Body | Function |
|---|---|---|
| Bulbospongiosus | Arises from perineal body (posterior attachment) | Compresses urethra/vagina; assists in erection; expels urine/semen. |
| Superficial Transverse Perineal | Inserts into perineal body (medial attachment) | Stabilizes perineal body; supports pelvic floor. |
| External Anal Sphincter | Anterior fibers attach to perineal body | Voluntary fecal continence. |
| Levator Ani (Puborectalis) | Some fibers insert into perineal body | Supports pelvic viscera; maintains anorectal angle. |
| Deep Transverse Perineal | Inserts into perineal body | Stabilizes perineal body; supports pelvic floor. |
| Rectovaginal/Rectourethral Septum | Attaches to superior aspect of perineal body | Separates rectum from vagina/urethra. |
The perineal body is the structural keystone of the perineum. Damage to the perineal body during childbirth (especially in 3rd and 4th degree tears) can lead to:
- Rectovaginal fistula - Abnormal communication between rectum and vagina.
- Fecal incontinence - Loss of external anal sphincter support.
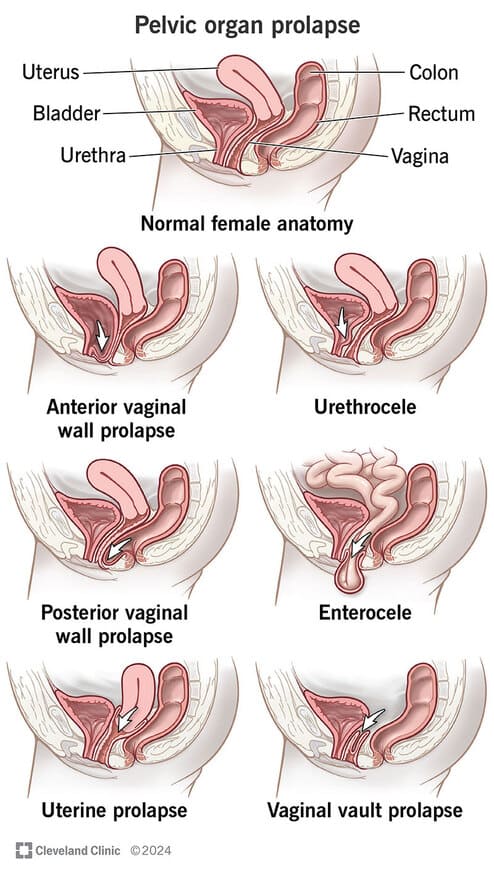
- Pelvic organ prolapse - Loss of central anchoring point for pelvic floor.
- Perineal descent - Bulging of the perineum during straining.
Surgical repair of the perineal body (perineorrhaphy) is essential after significant perineal tears to restore pelvic floor integrity.
SECTION 02: Fascial Layers of the Perineum
The perineum is organized into distinct fascial layers that create compartments, provide structural support, and define surgical planes. Understanding these layers is essential for surgery, regional anesthesia, and managing perineal trauma.
Superficial Perineal Fascia
The superficial perineal fascia in the urogenital triangle consists of two distinct layers:
Superficial Fatty Layer
The outer, more superficial layer of the perineal fascia:
- Continuous with Camper's fascia of the anterior abdominal wall.
- In females, forms the substance of the labia majora and the mons pubis.
- In males, largely replaced by the dartos muscle (smooth muscle of the scrotum).
- Contains fat and loose areolar tissue.
- Allows mobility of the skin over deeper structures.
Deep Membranous Layer (Colles' Fascia)
The deeper, more fibrous layer of the superficial perineal fascia:
- Lateral attachments: To the ischiopubic rami.
- Posterior attachment: To the posterior margin of the perineal membrane (and perineal body).
- Anterior continuity: With the dartos fascia of the penis/scrotum and Scarpa's fascia of the abdominal wall.
- Forms the floor of the superficial perineal space (pouch).
Colles' fascia is critical in containing urine extravasation from a ruptured spongy urethra. Because it is firmly attached to the ischiopubic rami laterally and the perineal membrane posteriorly, extravasated urine cannot spread into the thighs or anal triangle. Instead, it spreads:
- Anteriorly into the scrotum/penis (via continuity with dartos fascia).
- Superiorly onto the anterior abdominal wall (via continuity with Scarpa's fascia).
Perineal Membrane (Inferior Fascia of Urogenital Diaphragm)
A strong fibrous sheet stretching across the urogenital triangle, attached to the ischiopubic rami laterally and the perineal body posteriorly. It serves as the foundation for the external genitalia and divides the urogenital region into superficial and deep compartments. It was formerly called the "inferior fascia of the urogenital diaphragm."
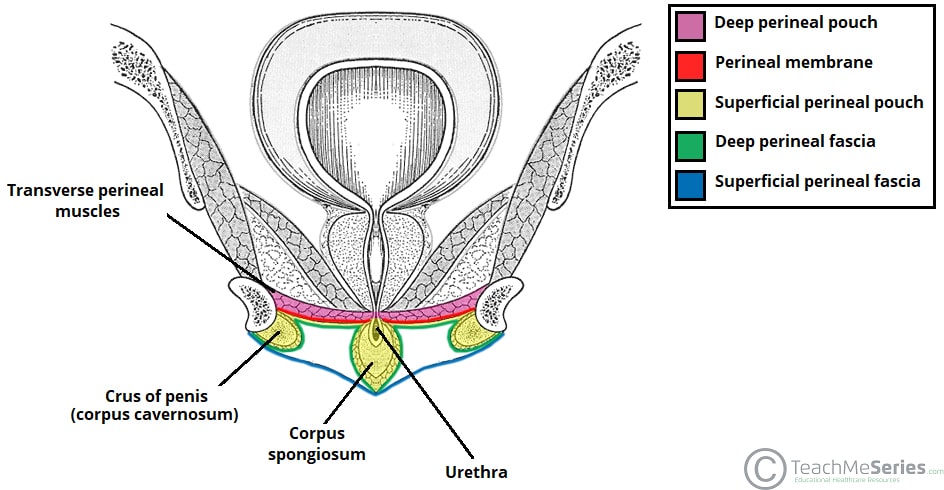
| Feature | Description |
|---|---|
| Attachments | Ischiopubic rami (lateral); perineal body (posterior); pubic symphysis (anterior). |
| Function | Supports external genitalia; divides urogenital triangle into superficial and deep spaces. |
| Clinical | Site of attachment for perineal muscles; barrier to infection spread. |
Deep Perineal Fascia (Gallaudet's Fascia)
A thin investing fascia that covers the superficial perineal muscles (ischiocavernosus, bulbospongiosus, and superficial transverse perineal). It lies deep to the superficial perineal fascia and invests the muscles of the superficial perineal pouch, providing a fascial sheath around each muscle.
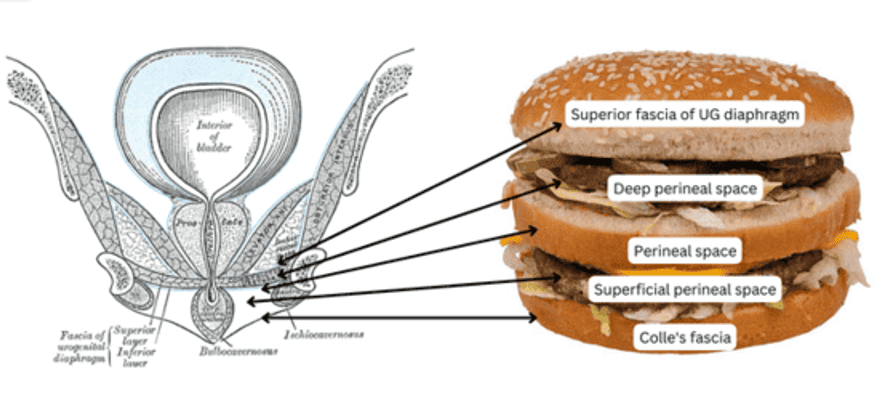
The "Burger" Model of Perineal Spaces
A helpful mnemonic for understanding the layered arrangement of the perineum:
- Superior fascia of urogenital diaphragm (pelvic diaphragm fascia) - Top Bun
- Deep perineal space (pouch) - Contains sphincter urethrae, deep transverse perineal - Meat Patty 1
- Perineal membrane - Middle Bun
- Superficial perineal space (pouch) - Contains erectile tissues, perineal muscles - Meat Patty 2
- Colles' fascia (deep membranous layer of superficial fascia) - Bottom Bun
SECTION 03: Urogenital Triangle: Compartments & Contents
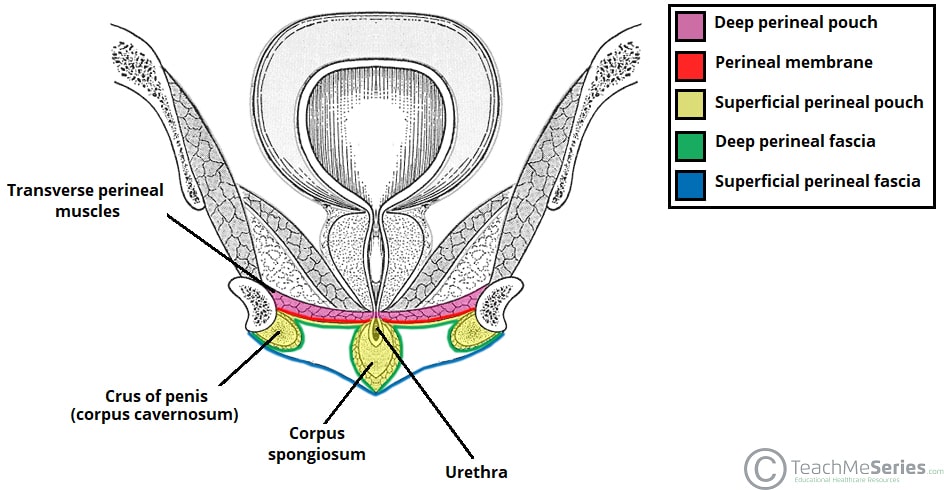
The urogenital triangle is divided by the perineal membrane into a superficial perineal space (pouch) and a deep perineal space (pouch). Each compartment contains distinct structures that are essential for urinary, reproductive, and sexual function.
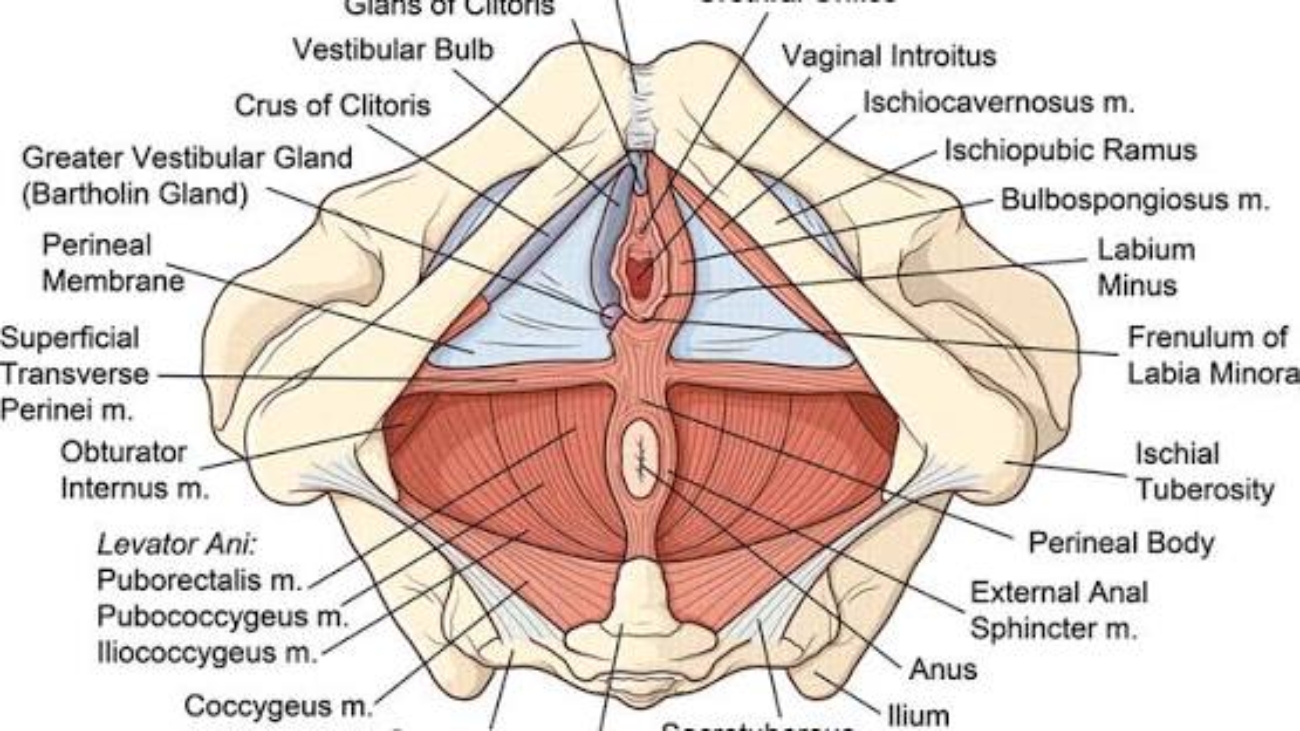
Superficial Perineal Space (Pouch)
The compartment between Colles' fascia (superficially) and the perineal membrane (deeply). It is bounded laterally by the ischiopubic rami and posteriorly by the perineal body. This space contains erectile tissues, muscles, vessels, and nerves.
Shared Structures
- Internal pudendal vessels - Terminal branches supplying the perineum.
- Branches of the pudendal nerve - Inferior rectal, perineal, and dorsal nerve branches.
- Perineal body - The central fibromuscular mass at the posterior boundary.
Muscles of the Superficial Perineal Space
Ischiocavernosus Muscle
Origin: Ischial tuberosity and ischial ramus.
Insertion: Crus of the penis/clitoris.
Innervation: Perineal branch of pudendal nerve.
Action: Compresses the crus, maintaining erection by restricting venous outflow.
Clinical: Essential for maintaining penile/clitoral erection. Weakness can contribute to erectile dysfunction.
Bulbospongiosus Muscle
Origin: Perineal body and midline raphe.
Insertion: Bulb of the penis (male) / Bulb of the vestibule (female).
Innervation: Perineal branch of pudendal nerve.
Action: Compresses urethra/vagina; expels urine/semen; assists in erection.
Clinical: In males, contraction after ejaculation expels residual semen from the urethra. In females, supports the vaginal orifice.
Superficial Transverse Perineal Muscle
Origin: Ischial tuberosity.
Insertion: Perineal body (medial).
Innervation: Perineal branch of pudendal nerve.
Action: Stabilizes the perineal body; supports the pelvic floor.
Clinical: Often absent or poorly developed; less functionally significant than other perineal muscles.
Sex-Specific Contents
| Male Contents | Female Contents |
|---|---|
| Root (bulb and crura) of the penis - The fixed, proximal portion of the penis attached to the perineal membrane and ischiopubic rami. | Clitoris (crura) - Erectile tissue attached to the ischiopubic rami. |
| Proximal spongy (penile) urethra - Passes through the bulb of the penis. | Bulbs of the vestibule - Paired erectile tissues on either side of the vaginal orifice. |
| Scrotal vessels and nerves - Posterior scrotal branches. | Greater vestibular glands (Bartholin's glands) - Open into the vestibule on either side of the vaginal orifice; secrete mucus for lubrication. |
| — | Labial vessels and nerves - Posterior labial branches. |
Deep Perineal Space (Pouch)
The compartment superior to the perineal membrane, bounded superiorly by the inferior fascia of the pelvic diaphragm (levator ani fascia). This space contains the membranous urethra, the external urethral sphincter, and associated vessels and nerves.
Shared Structures
- Membranous urethra - The shortest, least dilatable part of the male urethra (~1 cm); passes through the deep perineal space.
- Internal pudendal vessels - Terminal branches coursing through the space.
- Dorsal nerve of the penis/clitoris - Passes through to reach the dorsum of the erectile organ.
Muscles of the Deep Perineal Space
Sphincter Urethrae (External Urethral Sphincter)
Origin: Ischiopubic rami (medial aspect).
Insertion: Encircles the membranous urethra (male) or urethra and vagina (female).
Innervation: Perineal branch of pudendal nerve (somatic).
Action: Voluntary control of micturition; maintains urinary continence.
Clinical: Weakness causes stress urinary incontinence. In males, damage during prostatectomy can cause incontinence. In females, weakness is common after childbirth.
Deep Transverse Perineal Muscle
Origin: Ischial ramus.
Insertion: Perineal body (medial).
Innervation: Perineal branch of pudendal nerve.
Action: Stabilizes perineal body; supports pelvic floor.
Clinical: Often poorly developed; contributes to overall pelvic floor stability but is less clinically significant than the external urethral sphincter.
Male Unique Contents
- Bulbourethral Glands (Cowper's Glands): Paired glands located within the deep perineal space, posterolateral to the membranous urethra. Their ducts pierce the perineal membrane to enter the spongy urethra. They secrete a clear, viscous fluid that:
- Lubricates the urethra prior to ejaculation.
- Neutralizes acidic urine residue in the urethra.
- Contributes to the pre-ejaculate (pre-seminal fluid).
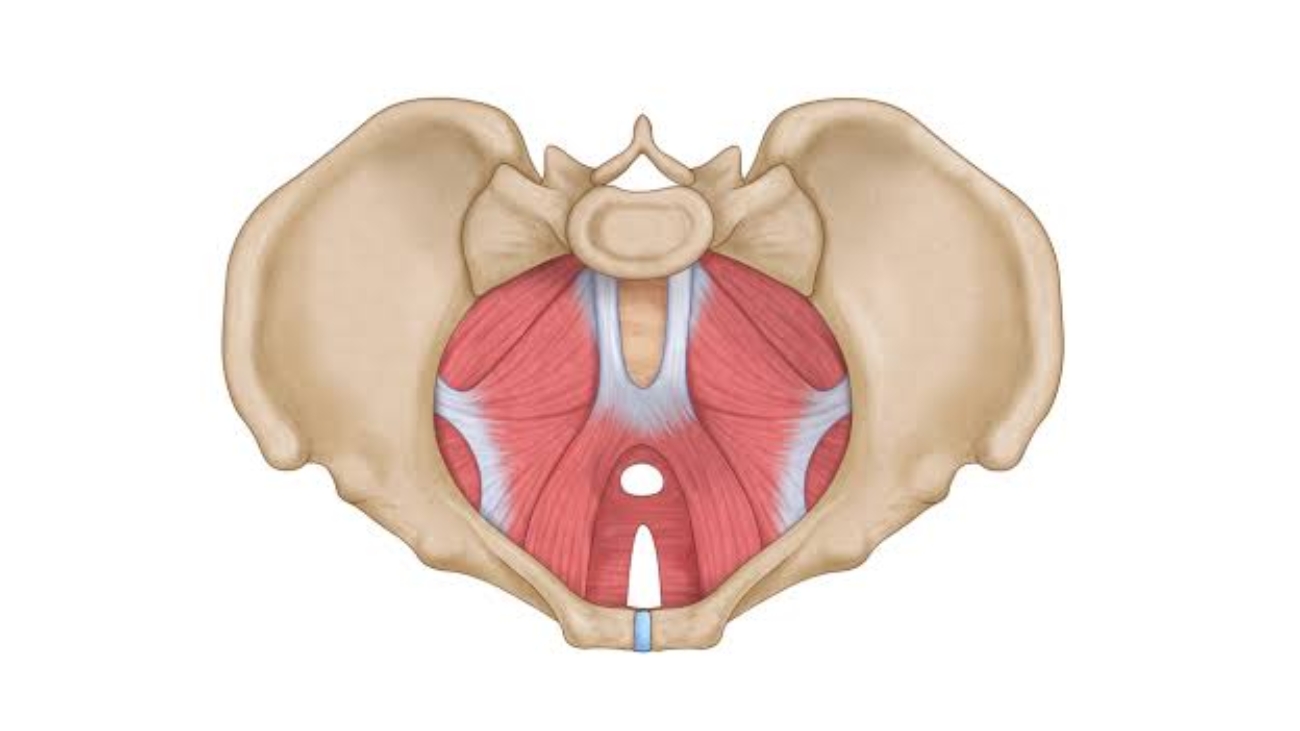
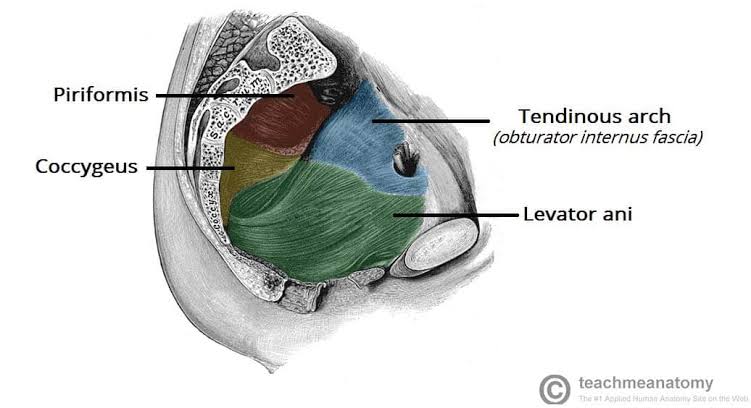
SECTION 04: Anal Triangle & Ischioanal Fossae
The anal triangle contains the anal canal, the external anal sphincter, and the paired ischioanal fossae — wedge-shaped spaces filled with fat that allow for expansion of the anal canal during defecation and accommodate the fetal head during childbirth.
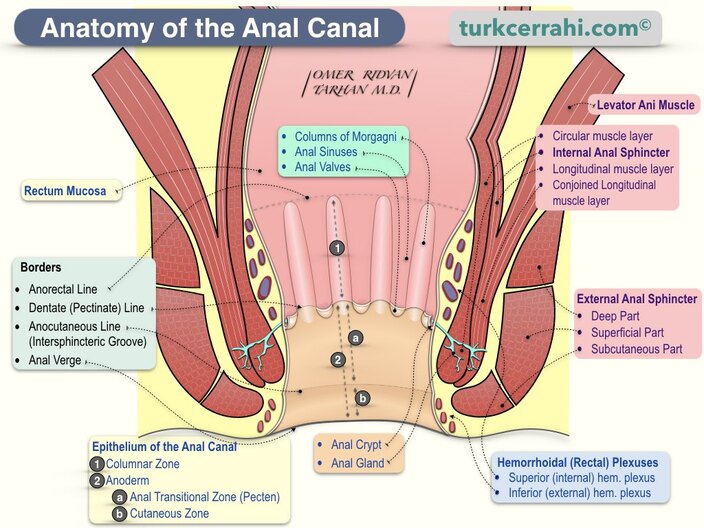
Anal Canal
The anal canal is the terminal portion of the alimentary tract, extending from the anorectal junction (where the rectum pierces the pelvic diaphragm) to the external opening (anus). It is approximately 3-4 cm long in adults.
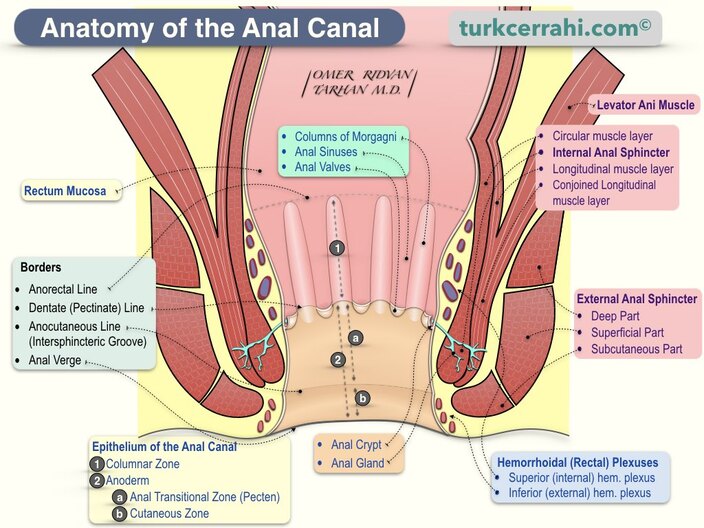
Sphincter Ani Externus (External Anal Sphincter)
The external anal sphincter is a skeletal muscle under voluntary control, composed of three parts:
| Part | Description | Innervation | Function |
|---|---|---|---|
| Subcutaneous | Most superficial; surrounds the anal orifice; no bony attachment. | Inferior rectal nerve (S2-S4) | Voluntary closure of anal orifice; maintains skin contact. |
| Superficial | Elliptical; attached to perineal body anteriorly and coccyx posteriorly. | Inferior rectal nerve (S2-S4) | Primary voluntary sphincter; provides squeeze pressure. |
| Deep | Circular; blends with puborectalis superiorly; no bony attachment. | Inferior rectal nerve (S2-S4) | Voluntary control; cooperates with puborectalis for continence. |
Control Mechanisms
The external anal sphincter is under voluntary somatic control via the inferior rectal nerve (a branch of the pudendal nerve, S2-S4). However, it also exhibits tonic involuntary activity at rest, maintaining continence without conscious effort. During defecation, it relaxes voluntarily while the puborectalis also relaxes, allowing the anorectal angle to straighten.
Ischioanal (Ischiorectal) Fossae
Large, wedge-shaped, fat-filled spaces that flank the anal canal laterally, one on each side. They are filled with adipose tissue and are of critical clinical importance as potential sites of infection and abscess formation.
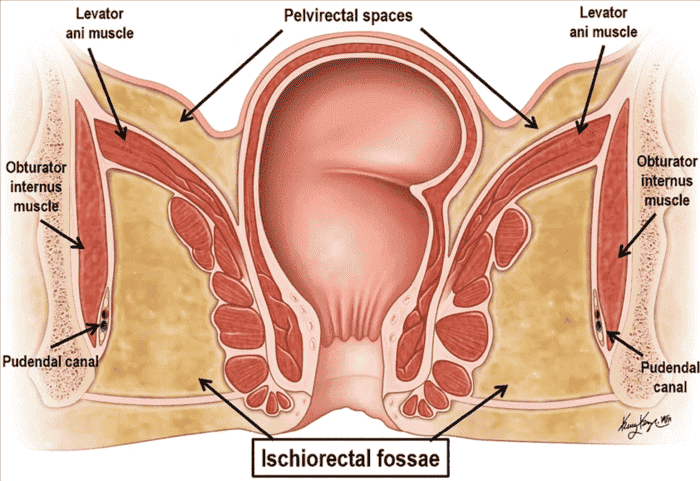
Boundaries of the Ischioanal Fossa
| Boundary | Structure | Clinical Relevance |
|---|---|---|
| Lateral Wall | Obturator internus muscle and ischial tuberosity | Site of pudendal canal (Alcock's canal) within obturator internus fascia. |
| Medial Wall | Levator ani muscle and external anal sphincter | Infection can spread to pelvic floor; surgical access to pelvic spaces. |
| Anterior Wall | Perineal membrane and transverse perineal muscles | Limits anterior spread of infection. |
| Posterior Wall | Gluteus maximus and sacrotuberous ligament | Infection can track posteriorly to contralateral fossa. |
| Apex | Junction of pelvic diaphragm and obturator fascia | Deep extension of abscesses. |
| Base | Skin of the perineum (perianal skin) | Site of perianal abscess drainage. |
Function of the Ischioanal Fossa
The fat-filled ischioanal fossae serve several important functions:
- Accommodate expansion of the anal canal during defecation.
- Allow passage of the fetal head during vaginal delivery.
- Support the pelvic floor by filling the space and providing cushioning.
- Allow pudendal neurovascular structures to course through the perineum.
The adipose tissue is highly vascular and innervated, making it susceptible to infection and pain.
Pudendal Canal (Alcock's Canal) within the Fossa
The pudendal canal (Alcock's canal) is a fascial tunnel located within the obturator internus fascia on the lateral wall of the ischioanal fossa. It houses:
- Pudendal nerve (S2-S4)
- Internal pudendal artery
- Internal pudendal vein
The canal runs from the lesser sciatic foramen anteriorly to the posterior border of the perineal membrane. It is the target for pudendal nerve block during vaginal delivery and perineal surgery.
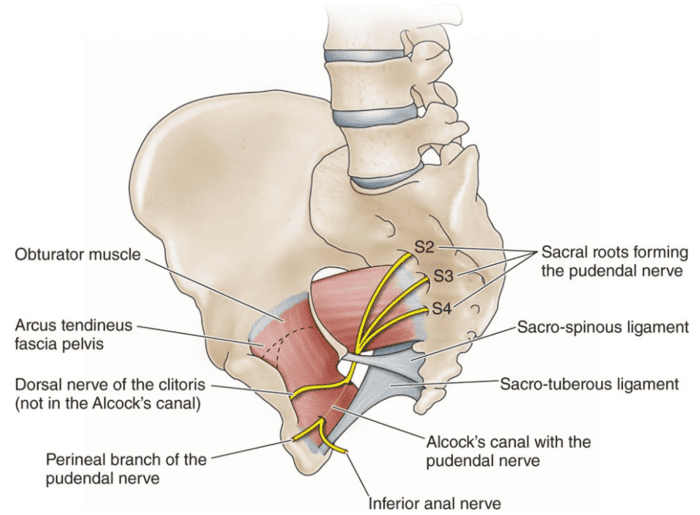
SECTION 05: Neurovascular Supply
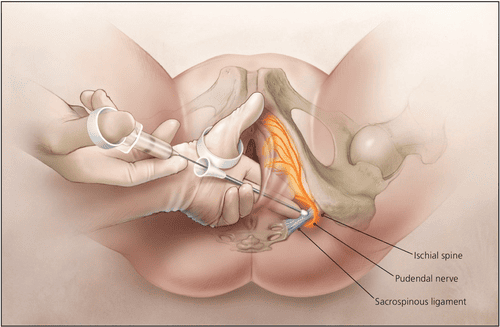
The perineum receives its blood supply from the internal pudendal artery and its branches, with innervation from the pudendal nerve (S2-S4). These structures follow a characteristic pathway from the pelvis, around the sacrospinous ligament, through the lesser sciatic foramen, and into the pudendal canal. Lymphatic drainage is divided between superficial inguinal nodes (for skin and external genitalia) and internal iliac nodes (for deep structures).
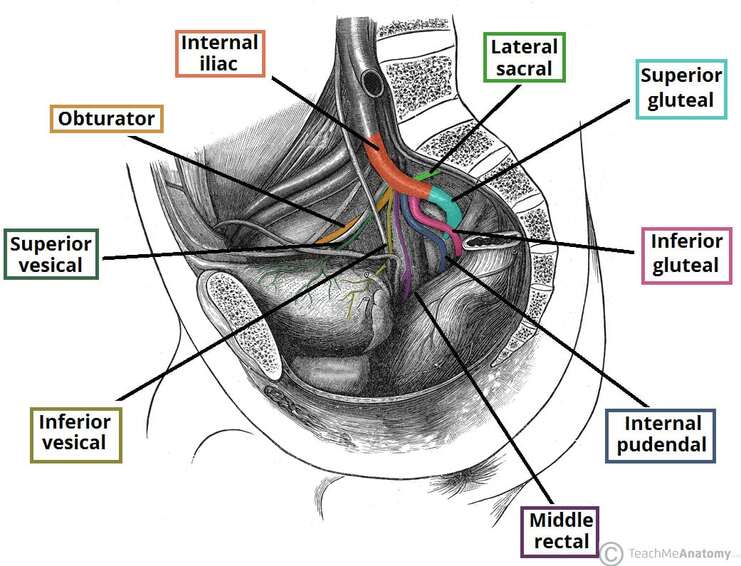
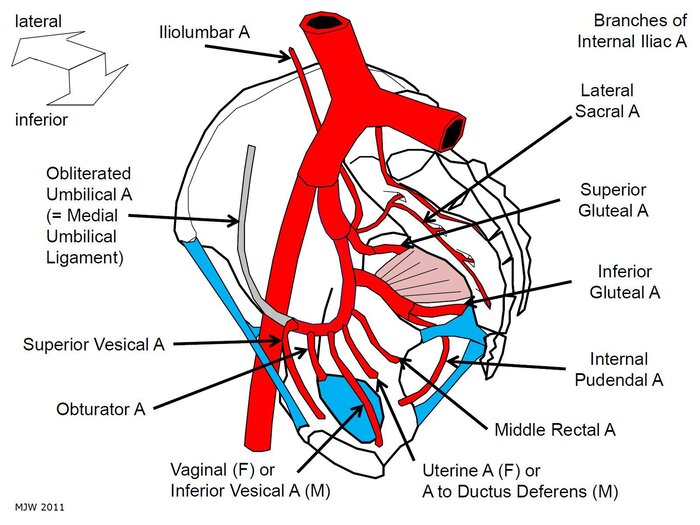
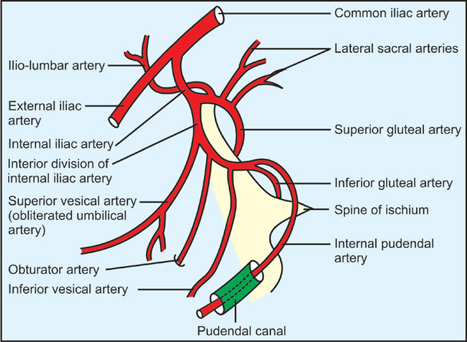
Internal Pudendal Artery
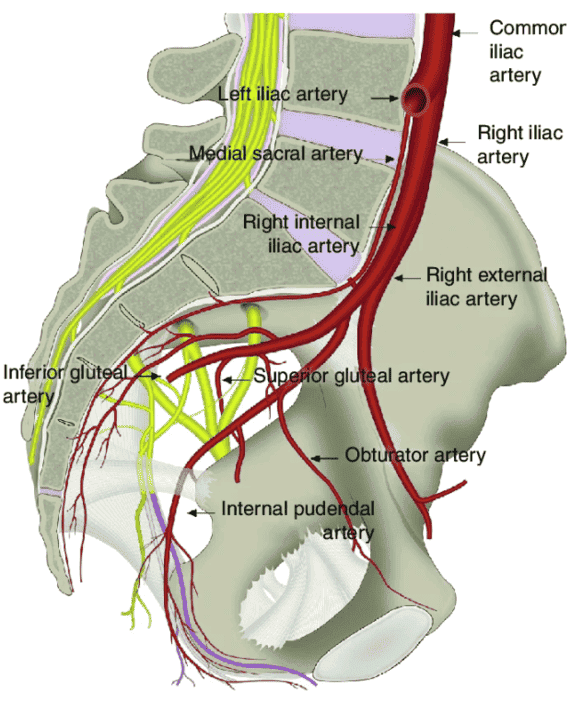
Course of the Internal Pudendal Artery
| Stage | Location | Key Landmark |
|---|---|---|
| Origin | Anterior division of internal iliac artery | Pelvic cavity, lateral to rectum. |
| Exit from Pelvis | Greater sciatic foramen (infrapiriform) | Below piriformis muscle. |
| Curve Around Ligament | Posterior to sacrospinous ligament and ischial spine | Palpable landmark for pudendal block. |
| Re-entry | Lesser sciatic foramen | Entering perineum. |
| Pudendal Canal | Within obturator internus fascia on lateral wall of ischioanal fossa | Alcock's canal. |
| Termination | Branches to perineum and external genitalia | See branches below. |
Branches of the Internal Pudendal Artery
| Branch | Distribution | Clinical Note |
|---|---|---|
| Inferior Rectal Artery | External anal sphincter, perianal skin, lower anal canal | Supplies below pectinate line; anastomoses with superior and middle rectal arteries. |
| Perineal Artery | Superficial perineal muscles, scrotum/labia, perineal skin | Gives off posterior scrotal/labial branches. |
| Artery of the Bulb | Bulb of penis/vestibule, bulbourethral glands | Penetrates perineal membrane. |
| Urethral Artery | Spongy urethra and corpus spongiosum | Runs within corpus spongiosum. |
| Deep Artery of the Penis/Clitoris | Corpus cavernosum of penis/clitoris | Primary artery for erection; runs within crus. |
| Dorsal Artery of the Penis/Clitoris | Dorsum of penis/clitoris, glans, skin | Runs with dorsal nerve; terminal branch of internal pudendal. |
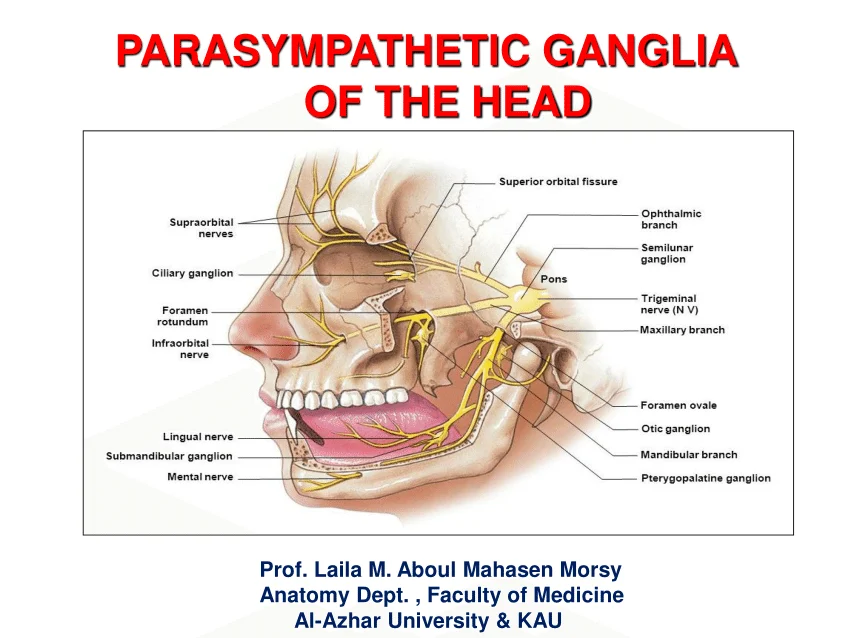
Pudendal Nerve (S2-S4)
The pudendal nerve follows the same pathway as the internal pudendal artery, exiting the pelvis via the greater sciatic foramen, passing around the sacrospinous ligament, and entering the perineum through the lesser sciatic foramen. It then courses through the pudendal canal (Alcock's canal) and gives off three primary branches:
1. Inferior Rectal Nerve
- Origin: Pudendal nerve within pudendal canal.
- Course: Passes medially across ischioanal fossa.
- Motor: External anal sphincter.
- Sensory: Perianal skin, lower anal canal (below pectinate line).
- Clinical: Damage causes fecal incontinence and loss of perianal sensation. The perianal "wink" reflex tests this nerve.
2. Perineal Nerve
- Origin: Pudendal nerve within pudendal canal.
- Course: Passes anteriorly into superficial perineal space.
- Motor: Superficial and deep perineal muscles (ischiocavernosus, bulbospongiosus, transverse perineal, sphincter urethrae).
- Sensory: Posterior scrotal/labial branches to skin of scrotum/labia majora.
- Clinical: Damage causes weakness of perineal muscles, urinary incontinence, and loss of scrotal/labial sensation.
3. Dorsal Nerve of the Penis/Clitoris
- Origin: Terminal branch of pudendal nerve.
- Course: Passes through deep perineal space, then runs along dorsum of penis/clitoris.
- Motor: None (purely sensory).
- Sensory: Skin of penis/clitoris, glans, prepuce.
- Clinical: Primary nerve for sexual sensation. Damage causes loss of sensation in the glans and erectile dysfunction. The dorsal nerve block is used for circumcision and penile surgery.
Lymphatic Drainage
The lymphatic drainage of the perineum is divided based on the tissue layer:
Skin & External Genitalia (Below the hymen/pelvic floor)
- Drains to superficial inguinal lymph nodes.
- Includes skin of perineum, scrotum, labia, penile skin, clitoral hood.
- Same drainage as lower limb skin.
- Clinical: Infections and cancers of the external genitalia (e.g., vulvar carcinoma, penile carcinoma) metastasize to superficial inguinal nodes.
Deep Structures & Internal Organs (Above the pelvic floor)
- Drains to internal iliac lymph nodes.
- Includes urethra, vagina (upper), prostate, anal canal (upper), bladder.
- May also drain to sacral lymph nodes.
- Clinical: Deep pelvic cancers (cervical, prostate, rectal) metastasize to internal iliac nodes before reaching inguinal nodes.
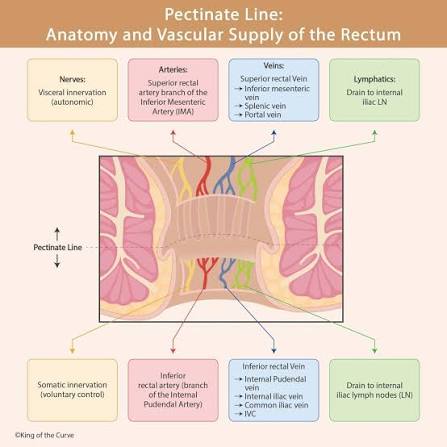
The Pectinate Line as a Lymphatic Boundary
The pectinate line of the anal canal serves as a critical lymphatic boundary:
- Above the pectinate line: Drains to internal iliac nodes (visceral drainage).
- Below the pectinate line: Drains to superficial inguinal nodes (somatic drainage).
This explains why anal canal cancers above the pectinate line metastasize to pelvic nodes first, while cancers below the line metastasize to inguinal nodes.
SECTION 06: Clinical & Applied Anatomy
The anatomy of the perineum has profound clinical implications in urology, obstetrics, gynecology, colorectal surgery, and emergency medicine. Understanding the fascial compartments, neurovascular pathways, and structural relationships is essential for managing trauma, infection, and childbirth complications.
Extravasation of Urine
A 35-year-old male sustains a straddle injury to the perineum. He presents with perineal swelling, bruising, and inability to void. A retrograde urethrogram reveals extravasation of contrast from the spongy urethra.
Mechanism: Straddle Injury | Site of Rupture: Spongy Urethra | Key Finding: Perineal Swelling
Anatomical Pathway of Extravasation:
- Urine escapes from the ruptured spongy urethra into the superficial perineal space.
- Because Colles' fascia is firmly attached to the ischiopubic rami laterally, urine cannot spread into the thighs.
- Because Colles' fascia is attached to the perineal membrane posteriorly, urine cannot spread into the anal triangle.
- Instead, urine spreads anteriorly into the scrotum (via continuity with dartos fascia) and superiorly onto the anterior abdominal wall (via continuity with Scarpa's fascia).
- This produces the characteristic "butterfly" perineal bruising and scrotal swelling.
Fascial Boundaries Contain the Spread
| Attachment | Effect |
|---|---|
| Lateral (ischiopubic rami) | Prevents spread into thighs. |
| Posterior (perineal membrane) | Prevents spread into anal triangle. |
| Anterior (dartos fascia of scrotum) | Allows spread into scrotum. |
| Superior (Scarpa's fascia of abdomen) | Allows spread onto anterior abdominal wall. |
Ischioanal Abscesses
Pathophysiology of Ischioanal Abscess
Infections originating in the anal canal (typically from anal glands) can spread into the fat-filled ischioanal fossa:
- Source: Infected anal gland (cryptoglandular origin) in the anal canal.
- Spread: Through the internal sphincter into the perianal space, then into the ischioanal fossa.
- Symptoms: Severe perianal pain, fever, swelling, fluctuance.
- Treatment: Surgical incision and drainage (I&D); antibiotics.
"Horseshoe" Abscess
Infections can track posteriorly from one ischioanal fossa to the other via the deep postanal space (the potential space posterior to the anorectal junction and anterior to the coccyx). This creates a "horseshoe" abscess that surrounds the anal canal:
- Pathway: Infection spreads from one fossa → deep postanal space → contralateral fossa.
- Clinical: Bilateral perianal swelling, severe pain, systemic signs of infection.
- Treatment: Requires drainage on both sides with a posterior counter-incision to break the "horseshoe."
Perineal Tears & Episiotomy
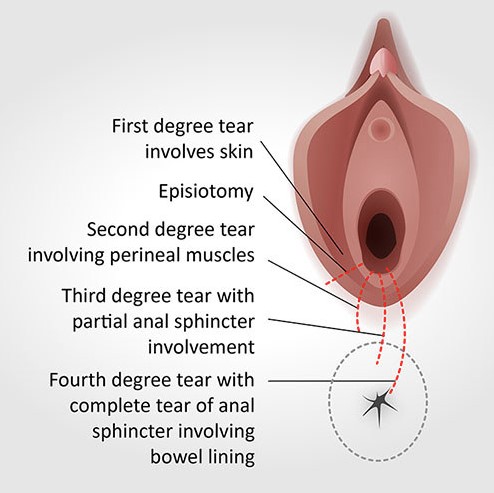
Perineal trauma during vaginal delivery is classified into four degrees based on the depth of tissue involvement:
| Degree | Structures Involved | Repair & Healing |
|---|---|---|
| 1st Degree | Skin and superficial perineal fascia only. | Repair: Simple suture. Healing: Excellent. |
| 2nd Degree | Perineal muscles involved (bulbospongiosus, transverse perineal). | Repair: Layered repair. Healing: Good with proper repair. |
| 3rd Degree | External anal sphincter torn. Subtypes: 3a (<50% thickness), 3b (>50% thickness), 3c (internal sphincter also torn). | Repair: Sphincter repair essential. |
| 4th Degree | Rectal mucosa involved (complete tear through sphincters into rectum). | Repair: Complex layered repair. Risk: Rectovaginal fistula, incontinence. |
Importance of Perineal Body Repair
The perineal body is the central structural anchor of the perineum. Preserving or surgically repairing the perineal body is critical to prevent:
- Fecal incontinence - Loss of external anal sphincter support.
- Rectovaginal fistula - Abnormal communication between rectum and vagina.
- Pelvic organ prolapse - Loss of central anchoring point for pelvic floor.
- Perineal descent - Bulging of the perineum during straining.
- Dyspareunia - Painful intercourse due to perineal scarring.
A mediolateral episiotomy (45 degrees from midline) is preferred over a median (midline) episiotomy because it directs the incision away from the perineal body and sphincter complex, reducing the risk of 3rd and 4th degree tears.
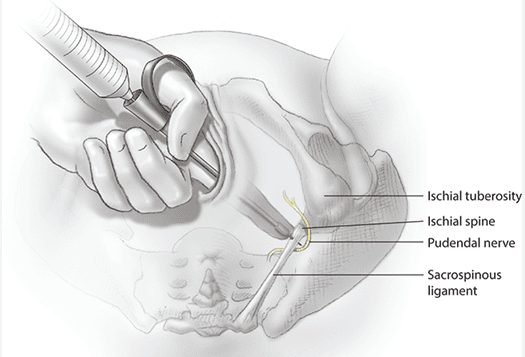
Pudendal Nerve Entrapment
A 42-year-old competitive cyclist presents with perineal numbness, erectile dysfunction, and pain during sitting. Symptoms worsen after long rides and improve with rest.
Condition: Pudendal Neuralgia | Mechanism: Nerve Compression | Common Name: Cyclist's Syndrome
Anatomical Mechanism of Compression:
- The pudendal nerve passes between the sacrotuberous ligament (posteriorly) and the sacrospinous ligament (anteriorly) as it exits the pelvis.
- Prolonged sitting (especially on narrow bicycle seats) compresses the nerve against these ligaments.
- The nerve is also compressed within Alcock's canal against the obturator internus fascia.
- Chronic compression leads to ischemia, demyelination, and axonal damage.
Clinical Manifestations:
- Perineal numbness - Loss of sensation in the perineum, scrotum/labia.
- Erectile dysfunction - Due to impaired parasympathetic vasodilation (S2-S4).
- Pain - Burning, aching, or stabbing pain in the perineum, worsened by sitting.
- Urinary symptoms - Frequency, urgency, dysuria.
- Defecatory pain - Pain during bowel movements.
Treatment: Behavioral modification (avoid prolonged sitting, use padded seats), physical therapy, pudendal nerve block, and in refractory cases, surgical decompression (transgluteal approach).