Major divisions of the Central Nervous System
A comprehensive note about the major divisions of the CNS
Module Learning Objectives & Overview
By the conclusion of this exhaustive master guide, you will be deeply conversant with:
- The Prosencephalon (Forebrain): Detailed anatomy of the Telencephalon (cortex, white matter tracts, basal ganglia) and the Diencephalon (thalamus, hypothalamus, epithalamus).
- The Mesencephalon (Midbrain): Structural division into the Tectum, Tegmentum, and Crus Cerebri, including cranial nerve exits.
- The Rhombencephalon (Hindbrain): In-depth exploration of the Metencephalon (Pons & Cerebellum) and Myelencephalon (Medulla Oblongata).
- The Spinal Cord: Gross external boundaries, internal gray/white matter organization, and ascending/descending tracts.
- The Ventricular System & CSF: Fluid production, circulation pathways, and clinical pathologies (Hydrocephalus).
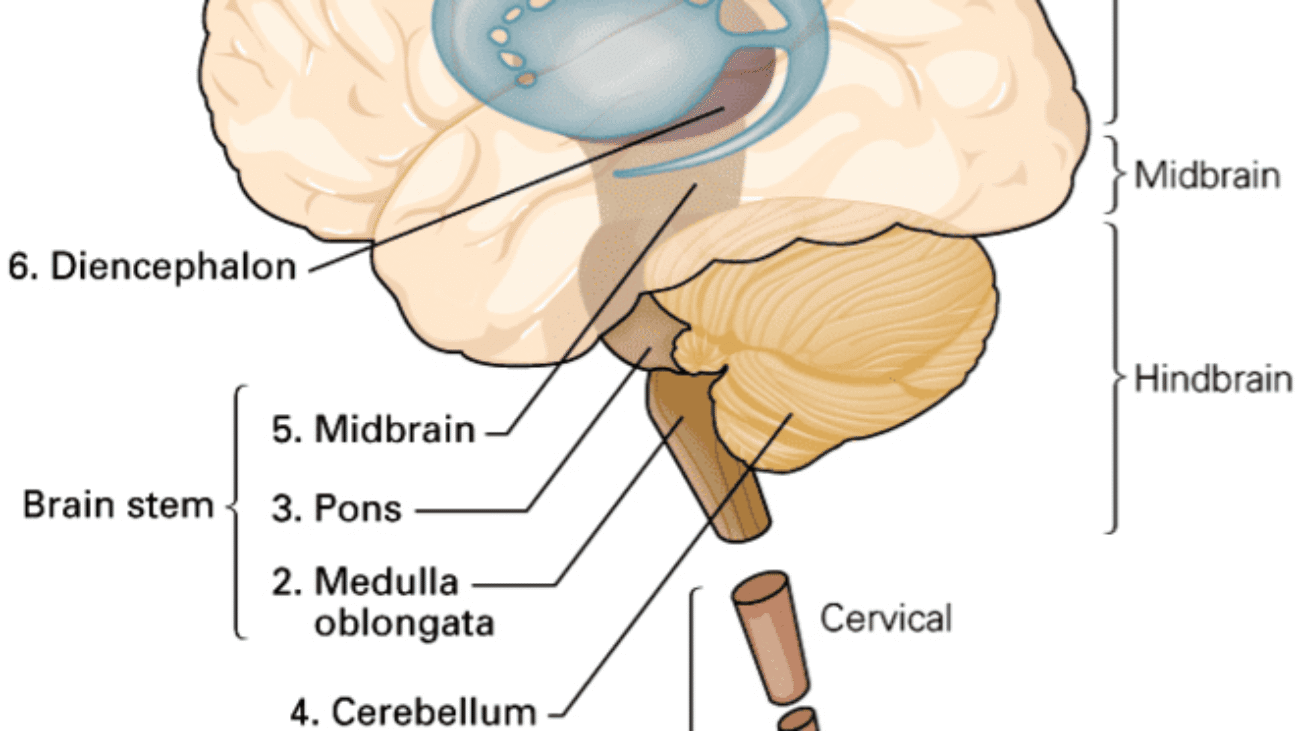
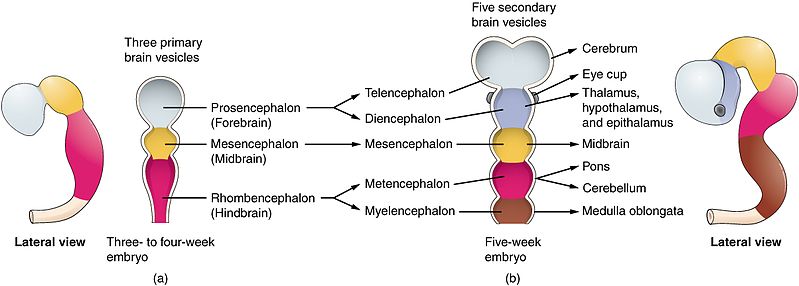
I. PROSENCEPHALON (Forebrain)
The prosencephalon, or forebrain, is the most rostral (forward) division of the developing neural tube. It is the seat of the highest level of neural processing in the human body. It gives rise to the telencephalon (which becomes the massive cerebral hemispheres and deep gray matter structures) and the diencephalon (the central core containing the thalamus, hypothalamus, and associated structures). Together, these regions process sophisticated sensory information, regulate primitive emotions, control fine voluntary movement, and coordinate higher cognitive functions like language, memory, and executive thought.
A. Telencephalon (Cerebral Hemispheres & Deep Structures)
The telencephalon forms the absolute largest part of the human brain. It consists of the two large cerebral hemispheres, highly convoluted surface cortex (gray matter), underlying massively connected white matter tracts, and deep gray matter nuclei (the basal ganglia and limbic structures).
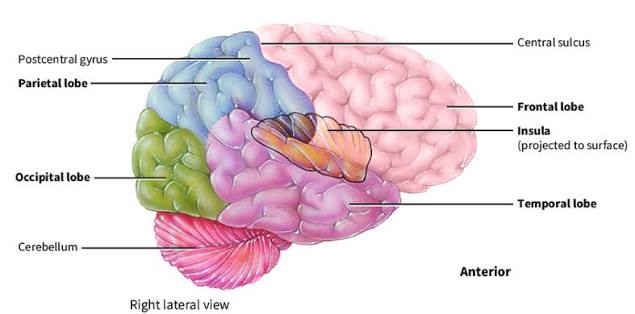
1. Surface Anatomy & Lobes
To fit the massive surface area of the cerebral cortex into the rigid skull, the brain folds into ridges called gyri and grooves called sulci (or deeper fissures). The cerebral cortex is divided into major lobes by three key anatomical landmarks:
- Central Sulcus (of Rolando): A prominent, continuous vertical groove separating the frontal lobe (anterior) from the parietal lobe (posterior). Clinical Importance: It is your absolute primary landmark for locating the primary motor and primary sensory cortices.
- Lateral Fissure (of Sylvius): A deep, sweeping horizontal fissure separating the temporal lobe (inferior) from the frontal and parietal lobes (superior). If you pull the lips (opercula) of this fissure apart, the hidden insular cortex lies buried deep within.
- Parieto-occipital Sulcus: Found primarily on the medial surface of the hemisphere, marking the definitive boundary between the parietal and occipital lobes.
Key Gyri to Master
- Precentral Gyrus: Located immediately anterior to the central sulcus. This is the primary motor cortex (Brodmann area 4). It houses the upper motor neurons that control voluntary movement of the contralateral (opposite) side of the body.
- Postcentral Gyrus: Located immediately posterior to the central sulcus. This is the primary somatosensory cortex (Brodmann areas 3, 1, 2). It receives all tactile, pain, temperature, and proprioceptive information from the contralateral body.
- Insular Cortex: A triangular-shaped lobe buried deep in the lateral fissure. It is highly involved in interoception (awareness of the body's internal state), taste processing (gustatory cortex), and primitive emotion/empathy.
EXAM TIP: On practical exams, always identify the central sulcus first to orient yourself.
Precentral = anterior = motor (Evolutionarily, action comes FIRST).
Postcentral = posterior = sensory (Perception of that action comes AFTER).
2. White Matter Tracts
White matter consists of heavily myelinated axon bundles connecting different brain regions. They are categorized into three types: Commissural fibers (connect hemispheres), Projection fibers (connect cortex to lower structures), and Association fibers (connect areas within the same hemisphere).
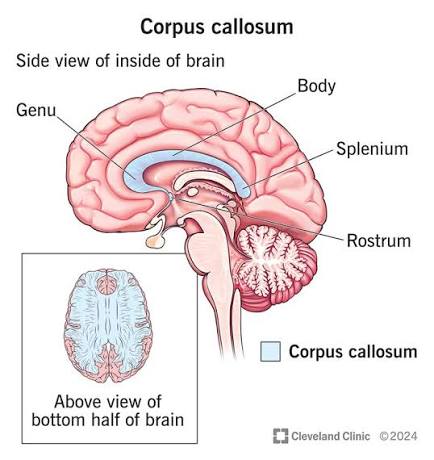
- Corpus Callosum: The absolute largest commissure in the brain, containing hundreds of millions of axons connecting the left and right cerebral hemispheres to ensure they act as a unified whole. It is divided into four parts (from anterior to posterior):
- Rostrum: The thin, beak-like anterior portion pointing downward.
- Genu: The sharp "knee" or bend at the anterior end.
- Body (Trunk): The long, massive central portion covering the lateral ventricles.
- Splenium: The thickened, bulbous posterior end (splenium means "bandage").
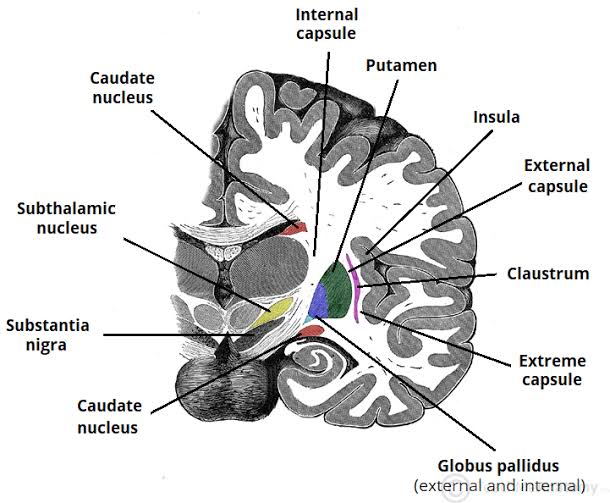
- Internal Capsule: A massive, V-shaped band of projection white matter containing almost all ascending (sensory) and descending (motor) fibers traveling between the cerebral cortex and subcortical structures (brainstem/spinal cord). It has three highly distinct parts:
- Anterior Limb: Located between the head of the caudate nucleus and the lentiform nucleus. Contains frontopontine and thalamocortical fibers.
- Genu: The sharp bend; critically contains corticobulbar fibers (the motor fibers descending to innervate the cranial nerve nuclei in the brainstem for face/jaw/swallow movement).
- Posterior Limb: Located between the thalamus and the lentiform nucleus. Highly critical as it contains the corticospinal tract (motor to the body) and major ascending somatosensory fibers from the thalamus to the cortex.
- Anterior Commissure: A smaller, secondary commissure connecting the temporal lobes and olfactory bulbs across the midline. It plays a role in pain sensation and smell.
- Fornix: A prominent C-shaped bundle of white matter that serves as the major output tract of the hippocampus, connecting it to the mammillary bodies of the hypothalamus. It is a critical highway for the limbic system and memory formation.
Clinical Pearl
Internal Capsule Stroke (Lacunar Infarct)
The internal capsule is incredibly HIGH-YIELD. Because millions of descending motor fibers are packed tightly into the tiny space of the posterior limb, a very small stroke here (a lacunar infarct caused by occlusion of the tiny lenticulostriate arteries) will cause massive, devastating pure motor hemiparesis (paralysis) on the contralateral side of the entire body. Rule: One small capsule, big consequences.
3. Deep Gray Matter (Basal Ganglia & Limbic Structures)
Deep within the white matter of the cerebral hemispheres lie clusters of neuronal cell bodies (gray matter) that regulate sophisticated aspects of movement, primitive emotion, and long-term memory.
- Corpus Striatum: The largest and most functionally critical component of the basal ganglia motor circuit. It consists of:
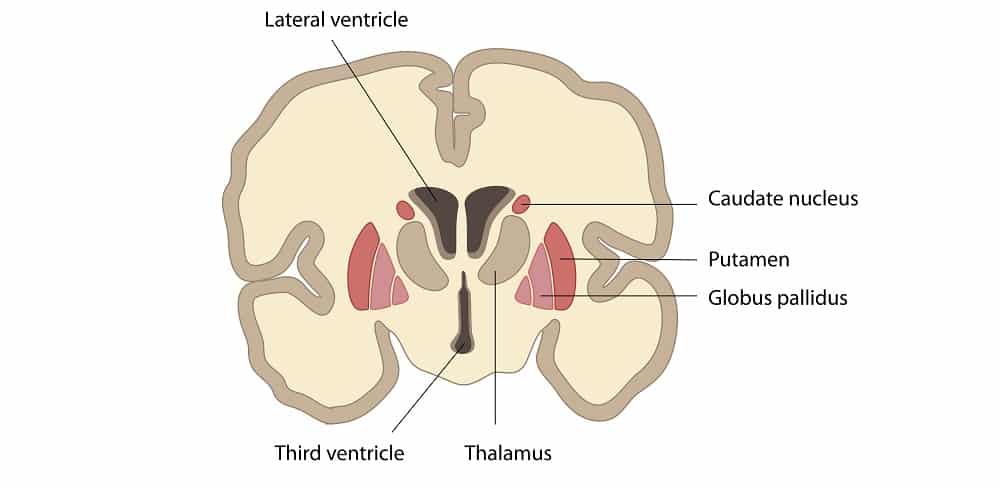
- Caudate Nucleus: A large, C-shaped structure that perfectly follows the curve of the lateral ventricle. It has three parts: the head (a large bulbous anterior portion bulging into the anterior horn of the lateral ventricle), the body (central portion), and the tail (tapering down and back into the temporal lobe).
- Lentiform Nucleus: A lens-shaped, wedge-like mass located lateral to the internal capsule. It is divided anatomically and functionally into:
- Putamen: The larger, darker, outermost lateral segment. It acts as the primary "receiver" of input from the cerebral cortex.
- Globus Pallidus: The smaller, paler, medial segment. Divided into external (GPe) and internal (GPi) segments. The GPi acts as the major inhibitory output nucleus of the basal ganglia, sending signals to the thalamus.
- Amygdaloid Nuclear Complex (Amygdala): An almond-shaped cluster of neurons located precisely at the tail of the caudate nucleus, buried deep within the medial temporal lobe. It is the brain's emotional threat center, processing fear, pleasure, aggression, and the emotional coding of memories.
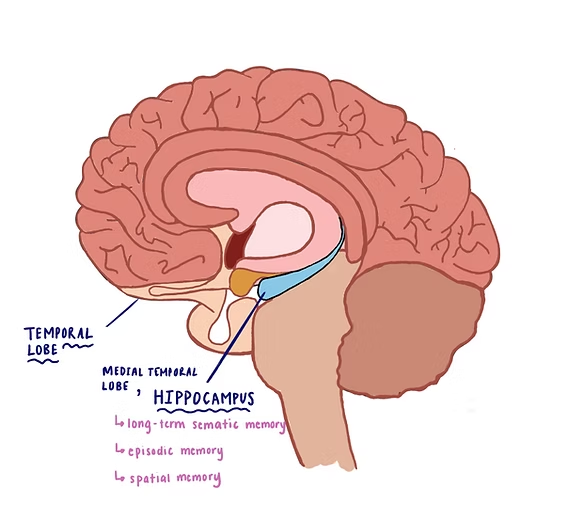
- Hippocampus & Dentate Gyrus: Located on the floor of the medial temporal lobe (specifically the inferior horn of the lateral ventricle). The hippocampus is the brain's "save button," responsible for converting short-term memories into permanent long-term memories and handling spatial navigation. The dentate gyrus is a heavily toothed, V-shaped strip of gray matter sitting atop the hippocampus, acting as the primary input gateway to the hippocampal formation.
EXAM TIP: Master this equation: Caudate + Putamen + Globus Pallidus = Corpus Striatum.
Furthermore: Putamen + Globus Pallidus = Lentiform Nucleus.
The caudate and putamen are physically separated by the white matter of the internal capsule, but they are functionally connected by tiny bridges of gray matter (giving the "striatum" its striped appearance).
Pathophysiology Expansion
Basal Ganglia Dysfunction
The basal ganglia do NOT directly initiate movement. Instead, they act as a quality control filter—they modulate, smooth out, and refine crude motor commands from the cortex. When this filter breaks, devastating movement disorders occur:
- Parkinson's Disease: Loss of dopaminergic input to the striatum leads to a rigid, hypokinetic state (resting tremor, stiff muscles, shuffling gait). The "go" pathway is broken.
- Huntington's Disease: Genetic degeneration of the GABAergic neurons specifically in the Caudate Nucleus leads to a hyperkinetic state (uncontrollable, dancing, choreiform movements). The "stop" pathway is broken.
B. Diencephalon (The Central Core)
The diencephalon sits deep in the absolute center of the brain, completely hidden beneath the corpus callosum and wedged between the two massive cerebral hemispheres. It acts as the ultimate relay and integration center for sensory, motor, and autonomic information. It consists of four distinct parts: the thalamus, hypothalamus, epithalamus, and subthalamus.
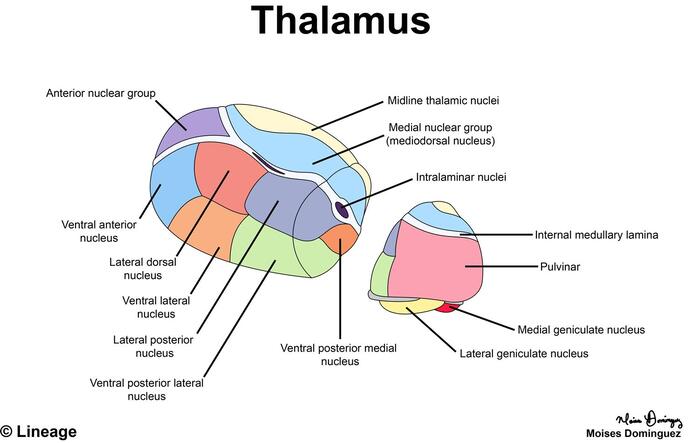
1. Thalamus
The thalamus is a massive, egg-shaped mass of gray matter that forms the bulk of the lateral wall of the third ventricle. It is universally known as the "gateway to the cortex" because virtually all sensory information (with the sole exception of olfaction/smell) must synapse here and be filtered before reaching the conscious cerebral cortex.
Key Features & Internal Structure:
- Interthalamic Adhesion (Massa Intermedia): A small, distinct bridge of gray matter crossing the midline straight through the third ventricle, physically connecting the left and right thalami. (Fun fact: It is absent in about 30% of normal human brains).
- Internal Medullary Lamina: A Y-shaped sheet of white matter inside the thalamus that anatomically divides it into three major nuclear tiers (Anterior, Medial, and Lateral).
- Nuclear Groups:
- Anterior Nuclear Group: Receives input from the mammillary bodies. Part of the Papez circuit (limbic system); heavily involved in memory consolidation and emotion.
- Medial Nuclear Group: Includes the mediodorsal (MD) nucleus; connects extensively to the prefrontal cortex; governs executive function, personality, and complex decision-making.
- Lateral Nuclear Group: The absolute largest group. Includes the critical sensory relay nuclei: the VPL (Ventral Posterolateral) for sensory input from the body, and the VPM (Ventral Posteromedial) for sensory input from the face.
- Pulvinar: The large, posterior expansion of the thalamus that overhangs the colliculi, involved in complex visual attention and integration.
- Medial Geniculate Body (MGB): Located on the posterior-inferior aspect; acts as the auditory relay center. It receives input from the inferior colliculus and projects to the primary auditory cortex in the temporal lobe.
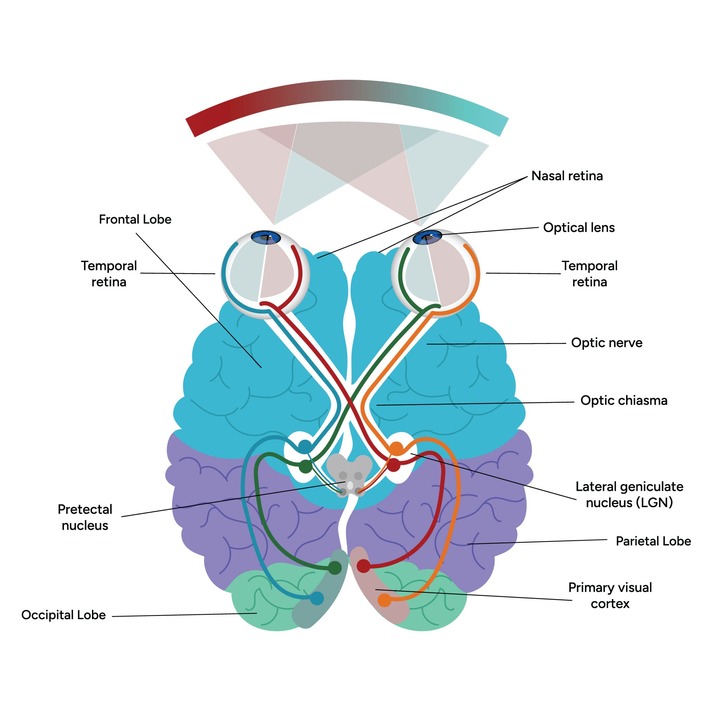
- Lateral Geniculate Body (LGB): Located slightly lateral and superior to the MGB; acts as the visual relay center. It receives input from the optic tract and projects massive optic radiations to the primary visual cortex in the occipital lobe.
EXAM TIP: Mnemonic to never forget the geniculate bodies:
Medial Geniculate = Music (Auditory relay).
Lateral Geniculate = Light (Visual relay).
Both sit on the posterior back of the thalamus like two small bumps. The LGB is lateral and slightly larger; the MGB is medial and smaller.
2. Hypothalamus
The hypothalamus is a small but incredibly mighty region located directly below the thalamus, forming the floor and lower lateral walls of the third ventricle. Despite weighing only about 4 grams, it is the master regulator of the body's internal environment (homeostasis). It controls body temperature, hunger, thirst, sleep-wake cycles, primitive emotions (rage/fear), and controls the entire endocrine system via the pituitary gland.
Key Surface Landmarks & Boundaries:
- Anterior Boundary: Marked by the Optic Chiasm (the X-shaped structure where half of the optic nerves cross over) and the lamina terminalis. The hypothalamus begins just above and behind this chiasm.
- Infundibulum (Pituitary Stalk): A thin, highly vascularized stalk connecting the base of the hypothalamus to the pituitary gland. It acts as a physical highway, carrying releasing hormones (via the hypophyseal portal blood system) to the anterior pituitary, and carrying direct axonal projections (from the supraoptic and paraventricular nuclei) down to the posterior pituitary to release Oxytocin and ADH.
- Posterior Boundary & Mammillary Bodies: Marked by two small, distinct round eminences on the ventral surface, located just posterior to the pituitary stalk. These Mammillary Bodies are part of the limbic system (Papez circuit) and are critical for memory. (Clinical Note: They are frequently destroyed in chronic alcoholics suffering from Wernicke-Korsakoff syndrome, leading to severe amnesia and confabulation).
- Superior Boundary: Separated from the massive thalamus above by a shallow groove called the Hypothalamic sulcus.
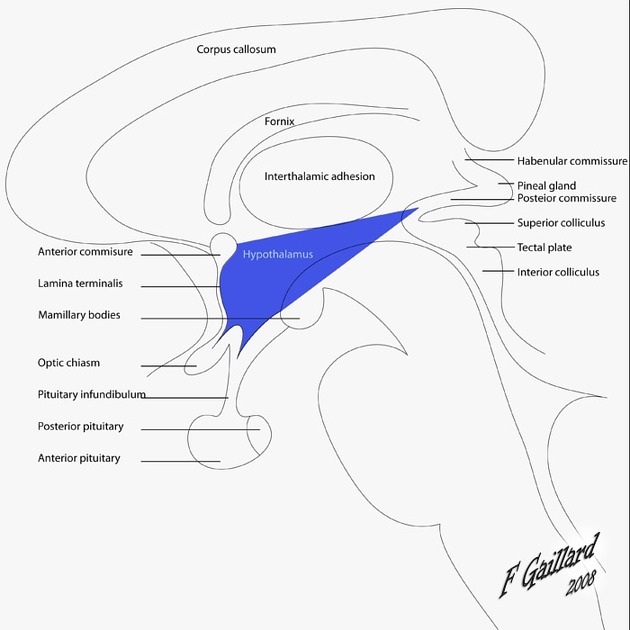
3. Epithalamus & Subthalamus
Epithalamus: The most dorsal (upper/posterior) part of the diencephalon, located posterior to the thalamus. It includes:
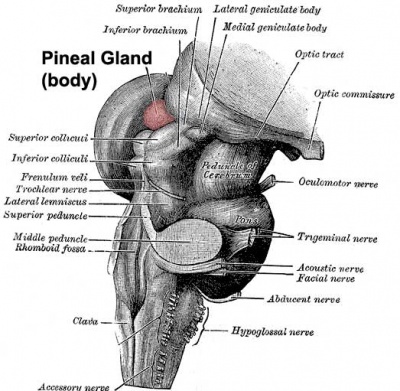
- Pineal Gland (Epiphysis): A small, pinecone-shaped endocrine gland that secretes melatonin, the hormone responsible for regulating circadian rhythms (sleep-wake cycles) based on light exposure. It sits precisely in the midline between the two superior colliculi and is attached to the posterior aspect of the third ventricle. (Clinical Note: In adults, it frequently calcifies and is used as a midline landmark on CT/MRI scans).
- Habenular Nuclei: Small nuclear clusters located in the habenular trigone just anterior to the pineal gland. They are involved in the limbic system, emotional processing, and reward pathways. They connect to the interpeduncular nucleus lower down via the habenulointerpeduncular tract (fasciculus retroflexus).
Subthalamus: Located ventral (below) the thalamus and lateral to the hypothalamus. It contains:
- Subthalamic Nucleus: A small, lens-shaped nucleus wedged tightly between the thalamus above and the internal capsule laterally. It is functionally a core part of the basal ganglia motor circuit (part of the indirect, movement-inhibiting pathway).
Clinical Application: Subthalamic Lesion
Because the subthalamic nucleus acts to suppress unwanted movements, a localized stroke (infarct) affecting this specific nucleus eliminates the "brakes" on the motor system. This results in a spectacular and highly distinctive clinical presentation known as Hemiballismus—characterized by violent, sudden, forceful, flinging movements of the arm and leg on the contralateral side of the body.
II. MESENCEPHALON (Midbrain)
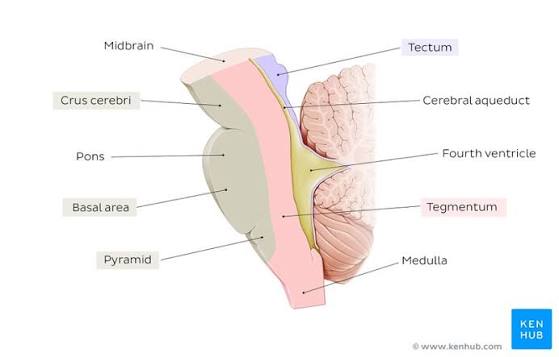
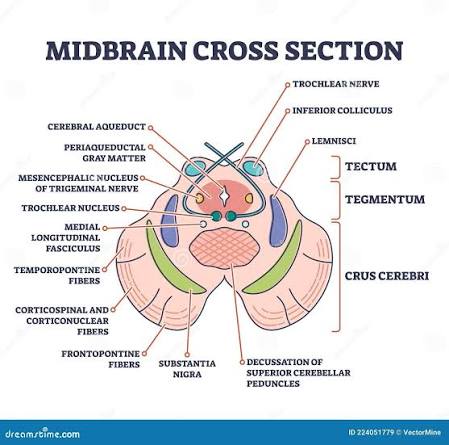
The mesencephalon, or midbrain, is the absolute shortest segment of the brainstem (measuring only about 2 cm in length). It acts as the critical bridge connecting the massive forebrain (diencephalon) above to the hindbrain (pons) below. Despite its tiny size, any damage here is catastrophic, as it contains critical structures for vision, hearing, gross motor movement, and the reticular activating system for basic arousal/consciousness. It is divided anatomically into three distinct layers from dorsal (back) to ventral (front): the tectum, tegmentum, and crus cerebri.
1. Tectum (Posterior Layer)
The tectum (Latin for "roof") is the dorsal-most part of the midbrain, located completely posterior to the fluid-filled cerebral aqueduct. It consists of four distinct rounded swellings (small hills) that collectively form the corpora quadrigemina (the "quadruplet bodies"):
- Superior Colliculi (paired, upper): These act as the brain's visual reflex centers. They receive direct input from the retina and visual cortex. They do not process conscious vision; rather, they rapidly coordinate unconscious eye movements and head-turning toward sudden visual stimuli (e.g., ducking when you see a baseball flying at your head out of the corner of your eye). Think of them as the "visual reflex headquarters."
- Inferior Colliculi (paired, lower): These act as auditory relay centers. They receive heavy input from the lateral lemniscus (the ascending auditory pathway) and send fibers up to the medial geniculate body of the thalamus. They are heavily involved in the startle reflex to loud noises and complex sound localization.
Brachia (The Connecting Arms):
- Brachium of the Superior Colliculus: A physical white matter arm that connects the superior colliculus to the lateral geniculate body (LGB) and the optic tract.
- Brachium of the Inferior Colliculus: A physical arm that connects the inferior colliculus to the medial geniculate body (MGB), ensuring the auditory signal reaches the thalamus.
EXAM TIP: Mnemonic to remember the corpora quadrigemina:
'Superior = Sight, Inferior = Sound.'
The superior colliculi are located higher up and handle visual reflexes; the inferior colliculi are lower down and handle auditory reflexes.
2. Tegmentum (Middle Layer)
The tegmentum (Latin for "covering") is the central, busy core of the midbrain. It is sandwiched firmly between the tectum (dorsal) and the substantia nigra/crus cerebri (ventral). It contains several vital descending tracts, ascending sensory pathways, and nuclear structures:
- Periaqueductal Gray (PAG): A prominent ring of gray matter immediately surrounding the cerebral aqueduct (of Sylvius). The PAG is highly rich in opioid receptors and endorphins. It is the brain's primary center for descending pain modulation (shutting down pain signals at the spinal cord level), defensive behavior, and vocalization.
- Red Nucleus: A massive, highly vascularized, pinkish nucleus located in the rostral (upper) midbrain tegmentum. It receives extensive input from the cerebellum and motor cortex, and sends a major motor tract (the rubrospinal tract) down to the spinal cord. This tract specifically facilitates flexor muscle tone of the upper limbs.
- Oculomotor Nerve (CN III): Motor rootlets for CN III emerge from the medial aspect of the cerebral peduncle, exiting directly into the interpeduncular fossa. CN III is critical for eye movement; it controls 4 of the 6 extraocular muscles, lifts the eyelid (levator palpebrae superioris), and carries parasympathetic fibers to constrict the pupil.
- Trochlear Nerve (CN IV): This is an incredibly unique nerve. It is the ONLY cranial nerve to exit from the dorsal (posterior) surface of the brainstem. It emerges just below the inferior colliculus, completely decussates (crosses over to the other side) inside the midbrain, and then wraps around the sides of the brainstem to reach the ventral side. CN IV innervates a single muscle: the superior oblique muscle (which depresses and intorts the eye).
EXAM TIP: CN IV (Trochlear) is biologically unique. Remember the "Rule of T":
'Trochlear = T = Two unique features.'
1. It has a Dorsal exit.
2. It undergoes complete Decussation before exiting.
3. Crus Cerebri & Ventral Layer
The crus cerebri (often referred to broadly as the cerebral peduncles) are the massive, prominent white matter pillars forming the ventral (anterior) surface of the midbrain. They are the major highways connecting the brain to the body.
- Substantia Nigra: A distinctive, darkly pigmented band of gray matter physically separating the tegmentum behind it from the crus cerebri in front of it. Functionally, it is a critical component of the basal ganglia. Its pars compacta region produces massive amounts of dopamine. The dark color is due to the accumulation of neuromelanin (a normal byproduct of dopamine synthesis).
Clinical Core: Severe, progressive degeneration and death of these dopaminergic neurons strips the basal ganglia of dopamine, causing the classic symptoms of Parkinson's disease (tremor, rigidity, bradykinesia). On autopsy, a Parkinson's brain lacks this dark black band.
- Crus Cerebri (Cerebral Peduncles): Two massive white matter stalks on the ventral midbrain. They contain literally millions of descending motor fibers from the cerebral cortex. The middle 3/5ths contain the corticospinal and corticobulbar tracts (the primary voluntary motor pathways). The outer edges contain corticopontine fibers traveling to the cerebellum.
- Interpeduncular Fossa: The deep, V-shaped groove located directly between the two massive cerebral peduncles on the ventral surface. As mentioned, CN III emerges directly from the walls of this fossa. The floor of this fossa is formed by the posterior perforated substance (pierced by tiny blood vessels).
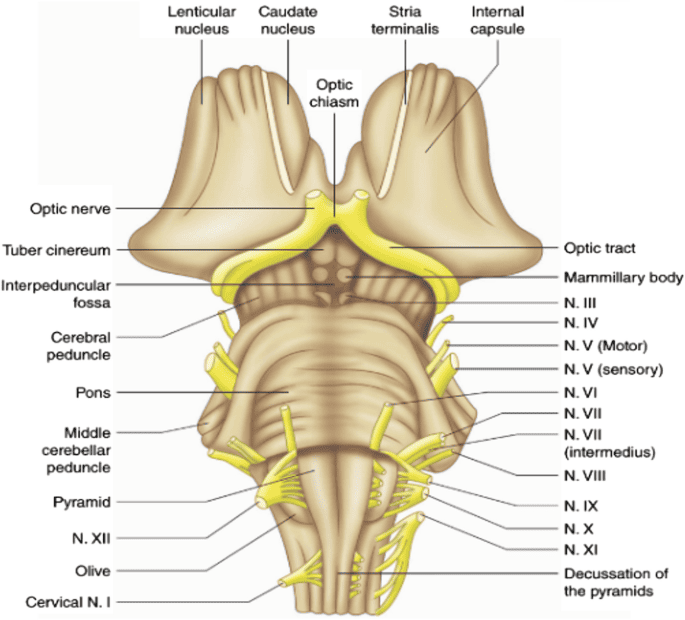
III. RHOMBENCEPHALON (Hindbrain)
The rhombencephalon, or hindbrain, is the most caudal (posterior/inferior) division of the developing embryonic brain. It connects the midbrain above to the spinal cord below. It consists of two major embryological parts: the metencephalon (which develops into the pons and cerebellum) and the myelencephalon (which develops into the medulla oblongata). The hindbrain is absolutely critical for basic survival, housing the autonomic centers that control subconscious breathing, resting heart rate, blood pressure tone, and highly coordinated balance/posture.
A. Metencephalon (Pons & Cerebellum)
1. The Pons
The pons (Latin for "bridge") is the prominent, bulging middle portion of the brainstem, wedged squarely between the midbrain and the medulla. It is approximately 2.5 cm long. Its massive anterior bulge is due to millions of transverse nerve fibers forming a literal "bridge" communicating between the cerebral cortex and the massive cerebellum behind it.
Anterior (Ventral) Surface Anatomy:
- Basilar Pons (Basis Pontis): The prominent, rounded anterior bulge. It contains descending longitudinal fibers (corticospinal tracts) diving down, interspersed with pontine nuclei. These nuclei receive signals from the cortex and send transverse pontocerebellar fibers across the midline and into the cerebellum, creating the massive horizontal striations visible on the surface.
- Basilar Groove (Sulcus): A distinct, shallow midline groove running vertically down the center of the anterior surface. It physically accommodates the massive basilar artery, a major blood vessel supplying the entire brainstem and cerebellum with oxygenated blood.
- Middle Cerebellar Peduncles (Brachium Pontis): The absolutely largest of the three cerebellar peduncles. They act as massive, thick columns extending laterally from the pons, plunging directly into the cerebellum. They exclusively carry the pontocerebellar INPUT fibers from the contralateral pontine nuclei into the cerebellar cortex, informing the cerebellum of what the motor cortex *intends* to do.
Cranial Nerve Exits of the Pons:
The pons is home to four critical cranial nerves (CN V through VIII).
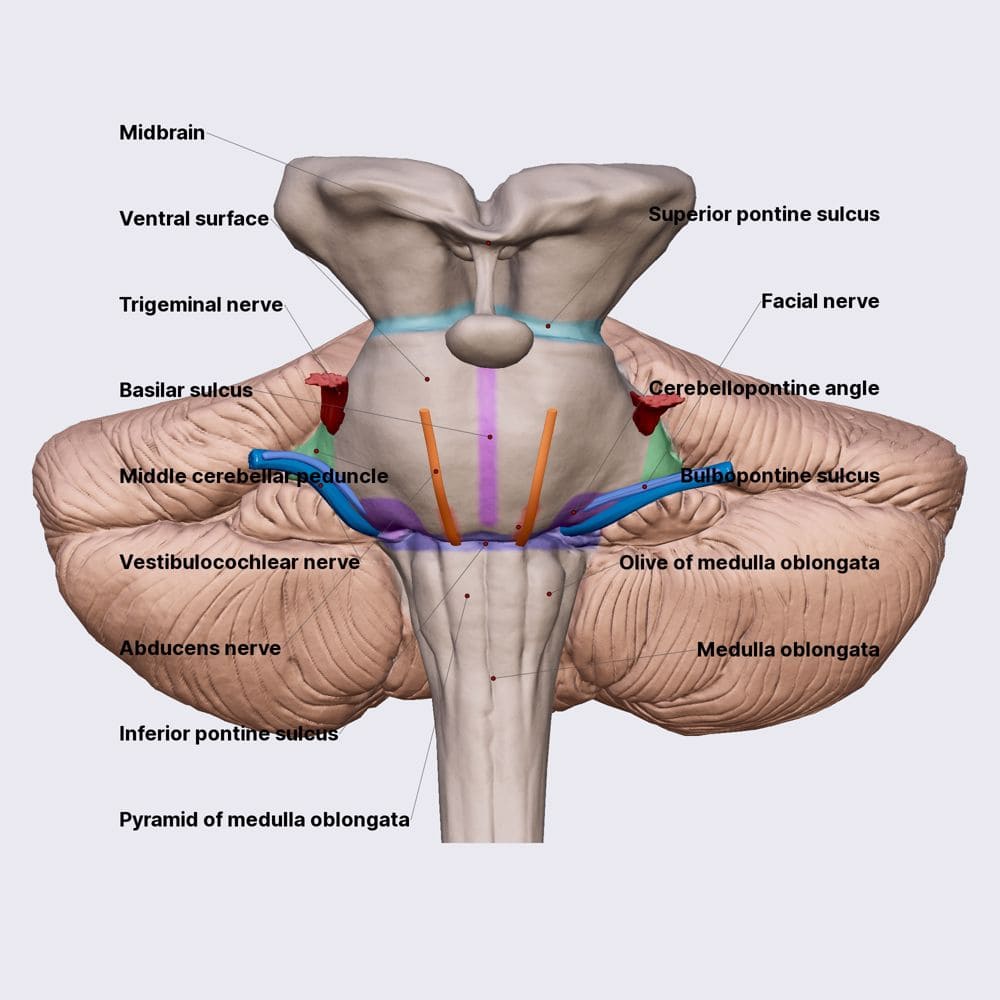
- Trigeminal Nerve (CN V): The largest cranial nerve. It emerges spectacularly from the mid-pons level on the anterolateral aspect. It has a large sensory root (carrying touch, pain, temp from the entire face) and a smaller motor root (innervating the muscles of mastication/chewing).
- Abducens (CN VI), Facial (CN VII), and Vestibulocochlear (CN VIII): These three nerves all emerge in a neat row at the pontomedullary junction (the deep horizontal groove separating the bottom of the pons from the top of the medulla).
- CN VI (Abducens) exits most medially, right above the medullary pyramids. It controls lateral eye movement.
- CN VII (Facial) and CN VIII (Vestibulocochlear) exit more laterally at the cerebellopontine angle (CPA).
Pathology Note
Cerebellopontine Angle (CPA) Tumors & Locked-In Syndrome
The CPA is a highly clinically important region. Slow-growing benign tumors called Acoustic Neuromas (Schwannomas) frequently grow on CN VIII here. As they expand, they compress CN VIII (causing unilateral hearing loss and ringing/tinnitus) and compress the adjacent CN VII (causing unilateral facial paralysis/drooping).
Furthermore, a massive stroke in the basilar artery can cause a catastrophic Pontine Infarct. Because the descending motor tracts (corticospinal) are destroyed bilaterally, but the sensory pathways and reticular activating system (consciousness) in the dorsal tegmentum are spared, the patient suffers from Locked-In Syndrome. The patient is fully awake, fully conscious, and feels everything, but is completely paralyzed from the neck down and cannot speak. They can often only communicate via vertical eye movements (controlled by the midbrain above the stroke).
2. The Cerebellum
The cerebellum (literally "little brain") sits posterior to the pons and medulla, tucked neatly beneath the occipital lobes of the cerebrum. It occupies almost the entirety of the posterior cranial fossa. While it does not initiate movement, it is absolutely critical for coordination, real-time error correction, balance, muscle tone, and motor learning (muscle memory like riding a bike). Astoundingly, it contains over 50% of all the neurons in the entire human brain, despite making up only 10% of the total brain volume!
Gross Divisions & Lobes:
- Vermis: The narrow, worm-like midline structure connecting the two massive cerebellar hemispheres. Functionally, the vermis controls the coordination and balance of the axial (trunk/core) musculature.
- Cerebellar Hemispheres: The two large, highly foliated lateral lobes. They functionally control the coordination, planning, and fine motor movements of the appendicular (limb) musculature.
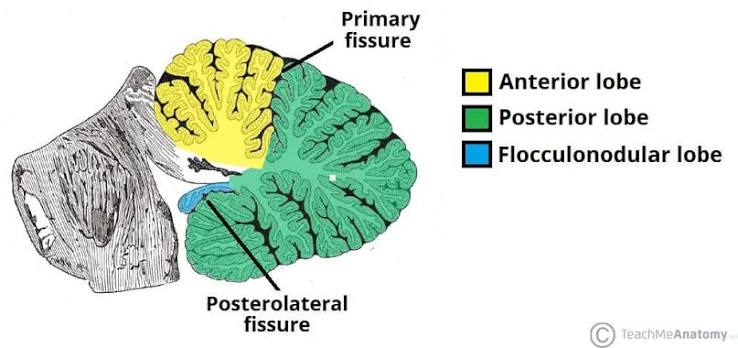
- Anterior Lobe: The superior/forward lobe. It is separated from the massive posterior lobe by the V-shaped primary fissure. It receives heavy proprioceptive (body position) input from the spinal cord via spinocerebellar tracts.
- Posterior Lobe: The absolute largest lobe. It is separated from the tiny flocculonodular lobe underneath by the posterolateral fissure. It receives massive input from the cerebral cortex (via the pons) to plan complex, skilled movements.
- Flocculonodular Lobe: The oldest and smallest lobe, consisting of the paired lateral flocculi and the central nodulus (part of the vermis). It is intimately wired to the vestibular system (inner ear) and controls equilibrium, balance, and complex eye movements.
Cerebellar Peduncles (The Three Connecting Bridges):
The cerebellum communicates with the brainstem exclusively through three paired bundles of white matter.
- Superior Cerebellar Peduncle (Brachium Conjunctivum): The primary OUTPUT pathway. It carries processed, corrective signals from the deep cerebellar nuclei up to the red nucleus and the thalamus, adjusting the motor commands.
- Middle Cerebellar Peduncle (Brachium Pontis): The primary INPUT pathway. It is the largest peduncle, carrying massive amounts of fibers from the contralateral pontine nuclei (bringing the "intent to move" plan from the cerebral cortex).
- Inferior Cerebellar Peduncle (Restiform Body): Carries a mix of BOTH input and output. It brings proprioceptive input from the spinal cord (spinocerebellar) and vestibular input from the inner ear, while sending output back to the vestibular nuclei to adjust balance.
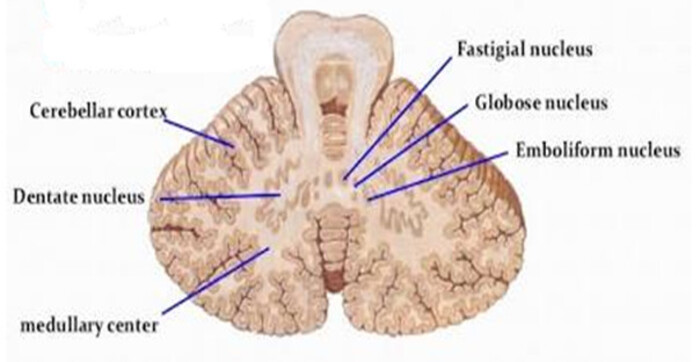
Deep Cerebellar Nuclei (Lateral to Medial):
The cerebellar cortex processes information, but the final output commands are generated by four pairs of deep gray matter nuclei buried inside the white matter core. From lateral (outside) to medial (center), remember the mnemonic:
'Don't Eat Greasy Food'
Dentate, Emboliform, Globose, Fastigial.
- Dentate Nucleus: The absolute largest, most lateral, and most heavily folded (looks like teeth) nucleus. It receives input from the lateral hemispheres and projects heavily to the thalamus to plan fine, skilled limb movements.
- Emboliform Nucleus: Receives input from the intermediate zone (paravermis); involved in adjusting ongoing limb movements.
- Globose Nucleus: Small, round, and located just medial to the emboliform; shares a similar function in limb coordination.
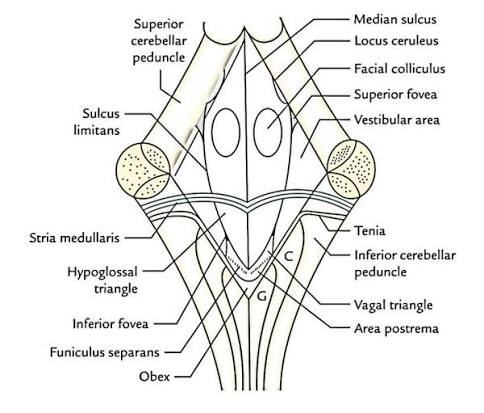
- Fastigial Nucleus: The smallest and most medial nucleus, sitting right in the roof of the 4th ventricle. It receives input from the vermis and flocculonodular lobe, projecting to vestibular nuclei to control upright posture, stance, and balance.
Clinical Application: Cerebellar Lesions
Unlike the cerebral cortex where a stroke causes paralysis on the opposite side, a cerebellar lesion causes profound uncoordinated movement (Ataxia) on the ipsilateral (SAME) side of the body. A patient with cerebellar damage will have a broad-based, staggering, "drunken" gait, and will exhibit an Intention Tremor (their hand will shake violently only when they try to reach out and touch a target, unlike Parkinson's where they shake at rest).
B. Myelencephalon (Medulla Oblongata)
The medulla oblongata is the most caudal (lowest) portion of the entire brainstem. It is continuous superiorly with the pons and inferiorly it transitions seamlessly into the spinal cord at the level of the foramen magnum (the large hole at the base of the skull). It is roughly 3 cm long. Functionally, it is the most critical structure for keeping you alive second-to-second; it houses the autonomic cardiovascular and respiratory centers controlling baseline breathing, heart rate, blood vessel tone, and primitive protective reflexes like vomiting, coughing, sneezing, and swallowing.
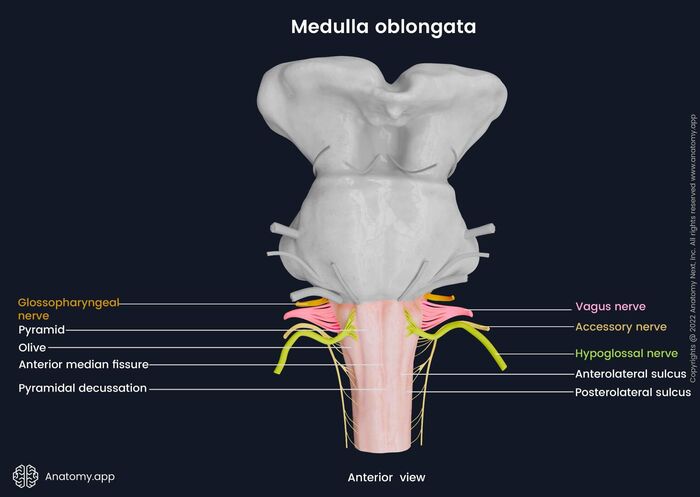
1. Anterior (Ventral) Surface Structures
The front of the medulla has several highly distinct morphological features:
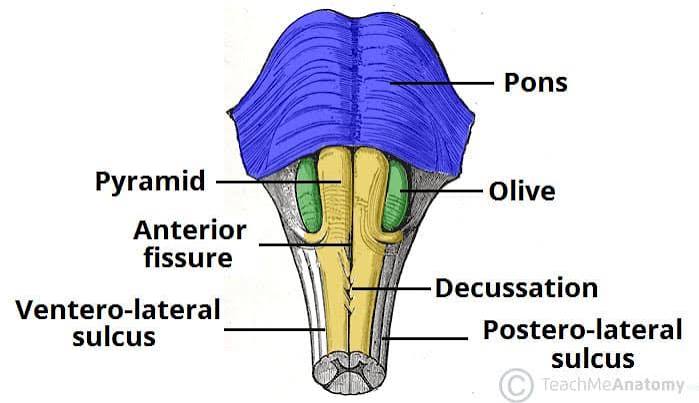
- Anterior Median Fissure: A deep, straight midline groove running down the ventral surface, perfectly continuous with the identical fissure on the front of the spinal cord. It cleanly separates the two pyramids.
- Pyramids: Two massive, smooth, longitudinal elevations sitting on either side of the anterior median fissure. They are named because they contain the descending corticospinal tract fibers (the "pyramidal tract") carrying voluntary motor commands from the motor cortex to the body.
Crucial Feature: At the absolute lowest, caudal end of the medulla, the anterior median fissure becomes obliterated. Here, approximately 85-90% of the massive corticospinal fibers plunge across the midline to the opposite side. This event is the pyramidal decussation. This physical crossing is the exact anatomical reason why a stroke in the left motor cortex paralyzes the right side of the body!
- Olives (Inferior Olivary Eminences): Prominent, smooth oval swellings located immediately lateral to the upper portion of the pyramids. They house the highly folded inferior olivary nuclei inside. These nuclei receive input from the spinal cord and red nucleus, and send millions of "climbing fibers" straight across into the contralateral cerebellum to drive severe motor learning and error correction.
Cranial Nerve Exits of the Medulla:
The medulla hosts the lowest four cranial nerves (CN IX through XII). Their exit points are defined entirely by the olive.
- Hypoglossal Nerve (CN XII): Emerges as several fine rootlets from the pre-olivary sulcus (the narrow vertical groove directly between the pyramid and the olive). It provides all motor control to the intrinsic and extrinsic muscles of the tongue.
- Glossopharyngeal (CN IX), Vagus (CN X), and Accessory (CN XI): All three of these nerves emerge in a vertical line from the post-olivary sulcus (the groove located behind the olive, between it and the inferior cerebellar peduncle).
- CN IX handles taste/sensation from the posterior 1/3 of the tongue and monitors blood pressure via the carotid sinus.
- CN X is the massive "wanderer," carrying 75% of the body's entire parasympathetic output to the thoracic and abdominal viscera (heart, lungs, gut).
- CN XI provides motor innervation to the sternocleidomastoid and trapezius muscles (allowing you to shrug shoulders and turn your head).
EXAM TIP: Medullary cranial nerves are anchored entirely by the olive.
PRE-olivary = Hypoglossal (CN XII). It sits in front.
POST-olivary = CN IX, X, XI. They sit in back.
The Pyramidal Decussation is one of the most highly tested anatomical facts in neuroanatomy because it defines contralateral motor control.
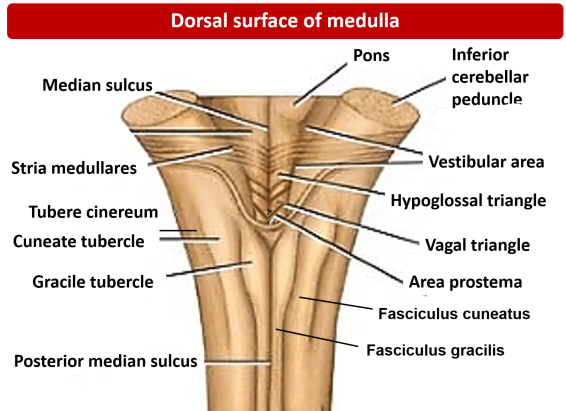
2. Posterior (Dorsal) Surface Structures
The posterior surface of the medulla looks completely different depending on whether you are looking at the lower half or the upper half. It is formally divided into two distinct regions: the closed medulla (inferior half) and the open medulla (superior half).
Closed Medulla (Inferior Half):
It is called "closed" because it still contains a tiny, fluid-filled central canal perfectly continuous with the central canal of the spinal cord below it. The dorsal surface features prominent sensory tracts ascending from the spinal cord:
- Posterior Median Sulcus: A shallow midline groove, continuing up from the spinal cord.
- Gracile Tubercle (Fasciculus Gracilis): The medial, rounded elevation right next to the midline. It contains the gracile fasciculus tract, which carries highly discriminatory fine touch, two-point discrimination, vibration, and conscious proprioception exclusively from the lower body (legs and lower trunk).
- Cuneate Tubercle (Fasciculus Cuneatus): A lateral, rounded elevation situated just outside the gracile tubercle. It carries the exact same fine touch/vibration modalities, but exclusively from the upper body (arms, upper trunk, neck).
- Posterior Intermediate Sulcus: A very fine groove physically separating the gracile and cuneate tubercles. It is incredibly important to note that this groove is present only above the T6 spinal level, because the cuneate tract (carrying arm sensation) only joins the spinal cord above T6!
Open Medulla (Superior Half):
It is called "open" because the narrow central canal suddenly flattens and splays wide open to form the lower, V-shaped half of the floor of the fourth ventricle (the rhomboid fossa).
Just below this open floor, the ascending sensory fibers in the gracile and cuneate tracts finally reach their first synapse at the gracile and cuneate nuclei (inside the tubercles). After synapsing here, the secondary neurons sweep forward and completely cross the midline (decussate) as the internal arcuate fibers. Once they cross, they ascend all the way up to the thalamus (VPL nucleus) as a thick ribbon called the medial lemniscus. This crossing is why the left side of the brain feels a feather touching your right toe!
Memory Hack
Never confuse the sensory dorsal columns:
Gracile starts with G = Ground (It carries sensation from the legs/lower body walking on the ground).
Cuneate starts with C = Ceiling (It carries sensation from the arms/upper body reaching for the ceiling).
IV. THE SPINAL CORD
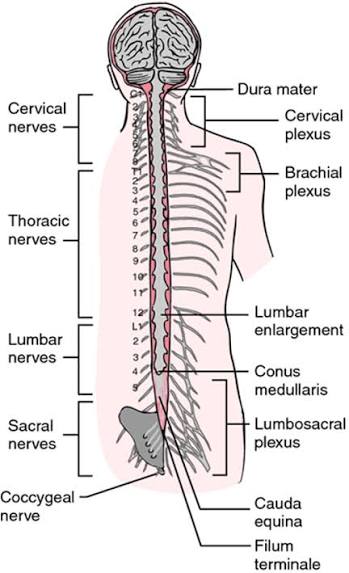
The spinal cord is a robust, somewhat flattened cylindrical column of dense nervous tissue. It extends from the medulla oblongata directly down through the bony vertebral canal. It serves as the massive, high-speed neural highway for bidirectional communication between the brain and the entire peripheral body, carrying powerful motor commands downward and transmitting continuous streams of sensory information upward. It also houses localized, independent reflex arcs (like the knee-jerk reflex) that protect the body faster than the brain can process.
1. Gross External Boundaries
- Superior Limit: The spinal cord begins precisely at the foramen magnum at the base of the skull, where it is flawlessly continuous with the medulla oblongata. Anatomically, this boundary is often defined just above where the first cervical spinal nerve (C1) rootlets exit.
- Inferior Limit (The Conus Medullaris): Unlike the vertebral column (bone), which continues growing through puberty, the spinal cord stops growing in length very early in childhood. Therefore, the adult spinal cord does not reach the bottom of the spine! It tapers to a sharp, cone-shaped termination called the conus medullaris. In the vast majority of healthy adults, the conus medullaris ends strictly at the L1-L2 vertebral body level (though it can occasionally range from T12 to L3).
Key Structures Below the Conus:
Because the spinal cord ends so high up, the lower lumbar, sacral, and coccygeal nerve roots must travel long distances downward inside the fluid-filled dural sac to reach their respective exit foramina in the lower spine.
- Cauda Equina ("Horse's Tail"): This massive bundle of descending nerve roots floating in CSF below the L2 level looks exactly like a horse's tail.
- Filum Terminale: A very thin, resilient, fibrous strand of pia mater tissue that extends from the very tip of the conus medullaris straight down to anchor the spinal cord. It prevents the cord from violently bouncing upward. It has two parts:
- Filum Terminale Internum: The portion floating within the dural sac, extending down to the S2 vertebral level where the dural sac ends.
- Filum Terminale Externum (Coccygeal Ligament): The portion that pierces through the dural sac, picking up a layer of dura and arachnoid, and continues down to bolt securely onto the coccyx (tailbone).
Spinal Cord Enlargements:
The spinal cord is not a uniform cylinder. It has two distinct, thick swellings located precisely where the massive bundles of nerves supplying the limbs enter and exit.
- Cervical Enlargement (C4-T1 spinal segments): This massive swelling gives rise to the brachial plexus, the complex nerve network that innervates the entire shoulder, arm, and hand.
- Lumbosacral Enlargement (L1-S3 spinal segments): This swelling gives rise to the lumbar and sacral plexuses, which innervate the heavy musculature of the pelvis, legs, and feet.
Clinical Pearl: The Lumbar Puncture (Spinal Tap)
Knowing that the solid spinal cord ends at L1-L2 in adults is clinically life-saving. When a doctor needs to extract CSF (to test for meningitis) or inject spinal anesthesia, they insert a long needle into the lower back. To absolutely guarantee they do not accidentally stab and permanently paralyze the solid spinal cord, the needle is ALWAYS inserted safely below L2—typically in the L3-L4 or L4-L5 intervertebral space. At this low level, the needle simply pushes into the Cauda Equina. The floating nerve roots effortlessly slide out of the needle's way, making the procedure highly safe!
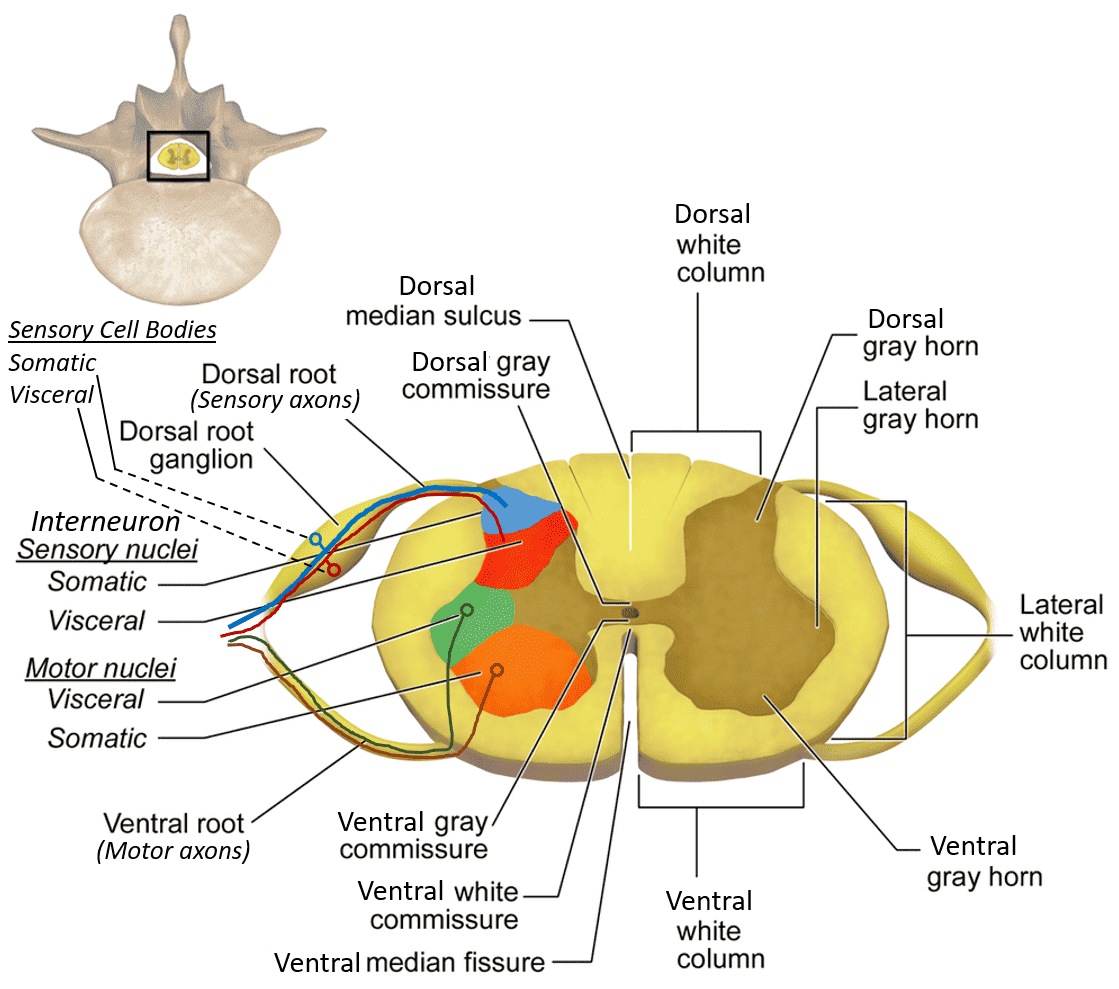
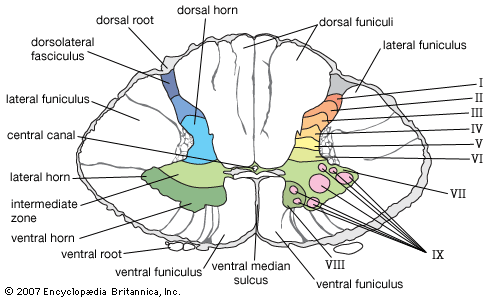
2. Internal Anatomy (Gray & White Matter)
A transverse cross-section of the spinal cord reveals a highly distinct, organized pattern: a central butterfly-shaped (or H-shaped) core of gray matter completely surrounded by a thick outer layer of white matter. This is the exact opposite of the cerebral cortex (where gray matter is on the outside).
Gray Matter (The Central Core):
The gray matter contains millions of unmyelinated neuronal cell bodies, dendrites, and synapses. It is the processing center. It is divided anatomically into three "horns":
- Dorsal (Posterior) Horn: The narrow, pointed back wing of the butterfly. It exclusively receives sensory input via the dorsal roots from the dorsal root ganglia outside the cord. It is packed with sensory interneurons and projection neurons that relay pain, temperature, and touch information up to the brain.
- Ventral (Anterior) Horn: The thick, broad front wing of the butterfly. It houses the massive cell bodies of the lower alpha motor neurons. Their axons shoot out of the cord via the ventral roots directly to the skeletal muscles to command voluntary contraction. (Polio and ALS specifically destroy these neurons, causing severe flaccid paralysis).
- Lateral Horn: A tiny, pointed lateral projection situated exactly between the dorsal and ventral horns. It is NOT present everywhere. It is found exclusively at specific spinal levels: T1-L2 (housing the sympathetic preganglionic neurons for the "fight or flight" response) and S2-S4 (housing the parasympathetic preganglionic neurons for pelvic organs like the bladder and bowels).
- Central Canal: A tiny, continuous fluid-filled channel located exactly in the center of the gray commissure (the bar connecting the two butterfly halves). It contains CSF and is continuous with the fourth ventricle high above in the brainstem.
White Matter (The Outer Highway):
The white matter completely surrounds the gray matter. It consists of massive, heavily myelinated axons organized into distinct bundles (tracts or fasciculi) traveling rapidly up or down the cord. It is topographically divided into three large columns, or funiculi:
- Anterior (Ventral) Funiculus: Located between the deep anterior median fissure and the exiting ventral horn roots. It primarily contains massive descending motor tracts (e.g., the anterior corticospinal tract for proximal muscle control, and the vestibulospinal tracts to keep you from falling over), along with some ascending crude touch fibers (anterior spinothalamic).
- Lateral Funiculus: Wedged between the dorsal and ventral horns. This is the busiest highway in the cord. It contains the absolutely critical lateral corticospinal tract (the main descending motor highway for precise, voluntary limb movement), the lateral spinothalamic tract (the main ascending sensory highway for sharp pain and extreme temperature), and the massive dorsal spinocerebellar tracts carrying unconscious joint position to the cerebellum.
- Posterior (Dorsal) Funiculus: Located entirely behind the dorsal horns. This is an exclusively sensory ascending highway. As previously discussed in the medulla section, it contains the fasciculus gracilis (the medial bundle carrying fine touch/vibration strictly from the lower body) and the fasciculus cuneatus (the lateral bundle carrying the same modalities strictly from the upper body).
EXAM TIP: A fast way to remember the white matter organization:
Anterior = Massive Motor commands going down.
Posterior = Highly specific fine Sensation coming up (gracilis for legs, cuneatus for arms).
Lateral = A chaotic mix of both vital motor (corticospinal) and vital pain/temp sensation (spinothalamic).
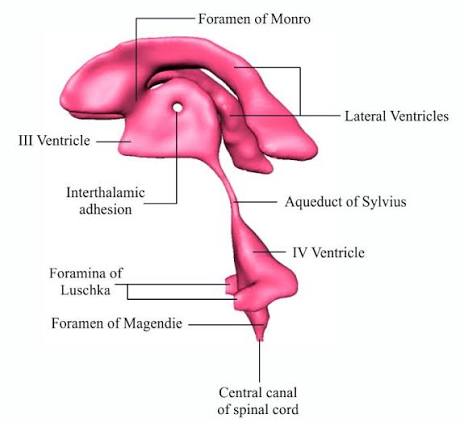
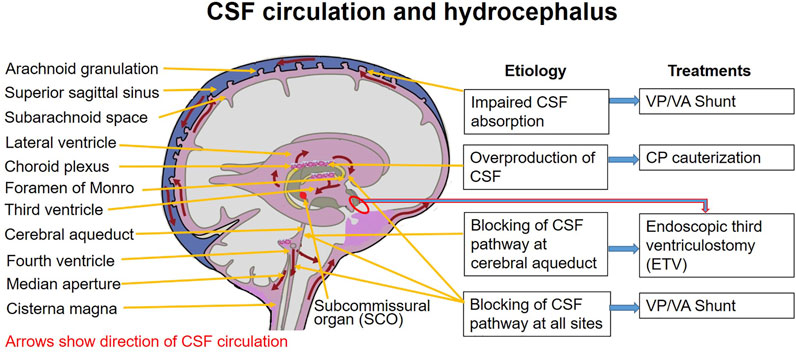
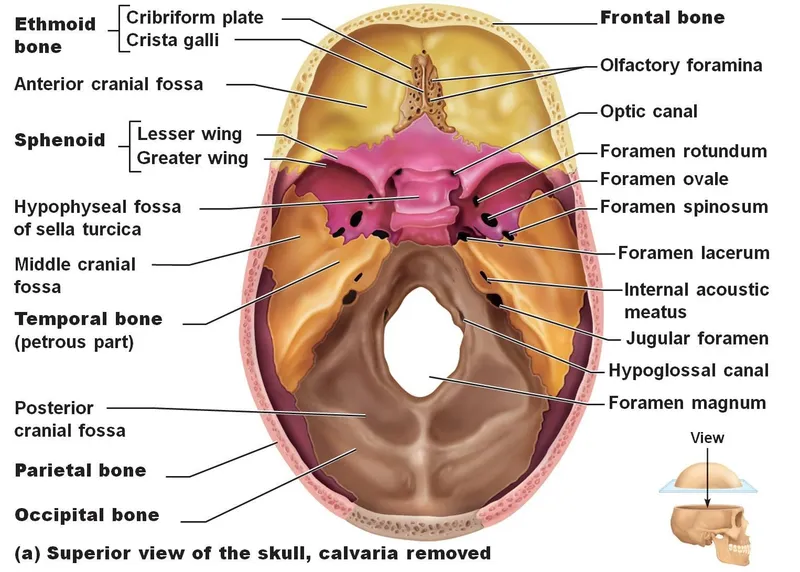
V. VENTRICULAR SYSTEM & CSF ANATOMY
The human brain is incredibly soft and heavy (weighing about 1.5 kg). If it rested directly on the hard skull bone, its own weight would crush its inferior vessels and neurons. To prevent this, the brain literally floats. The ventricular system is an intricate set of four interconnected, fluid-filled cavities hidden deep within the brain parenchyma. These cavities continuously produce, circulate, and resorb cerebrospinal fluid (CSF). CSF acts as a physical shock absorber, cushions the brain during trauma, removes toxic metabolic waste, and maintains a perfectly stable, specialized chemical environment required for delicate neural tissue to fire correctly.
1. Ventricles & Complex Connections
The system is laid out chronologically from the top of the brain down to the spinal cord:
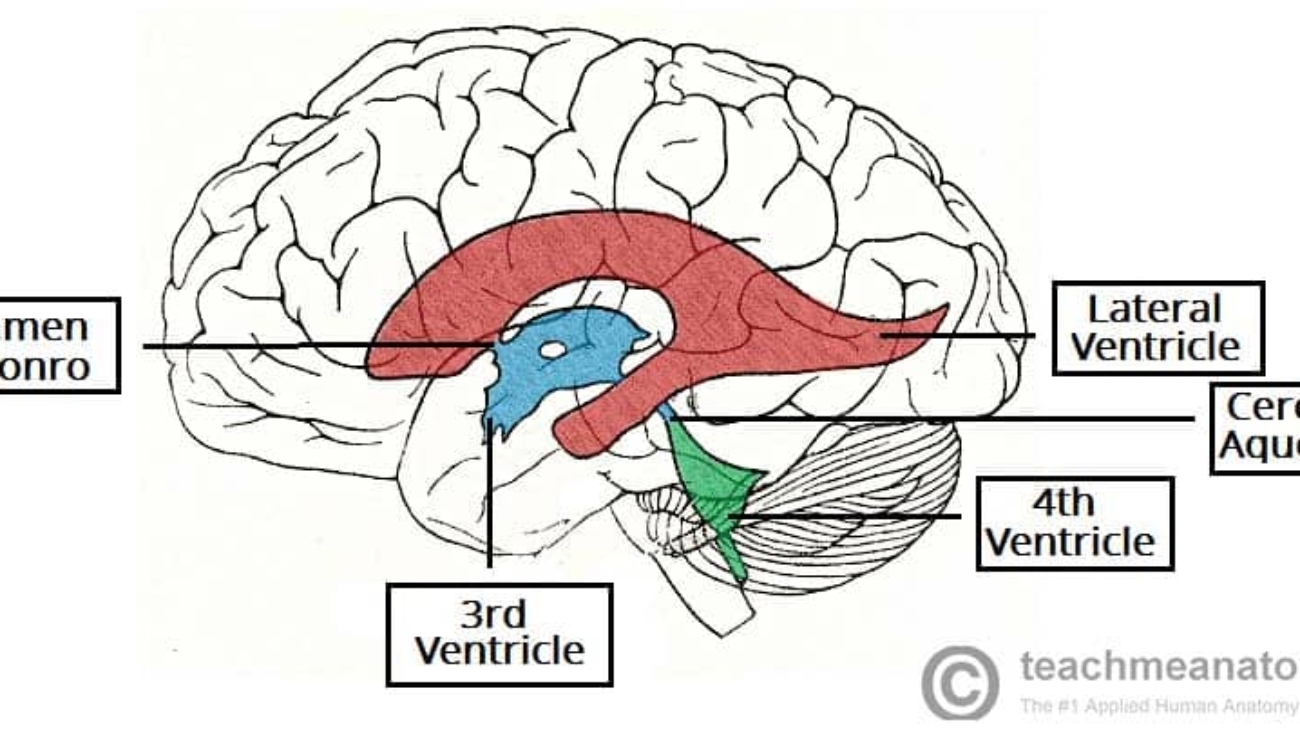
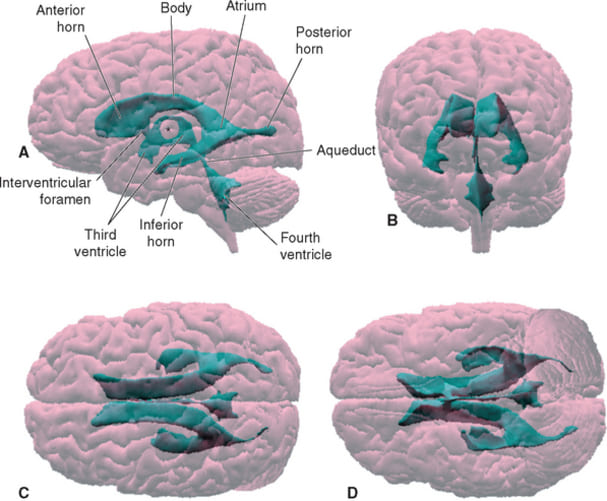
- 1 & 2. Lateral Ventricles (Paired): These are the absolute largest ventricles, forming massive C-shaped lakes buried deep within each cerebral hemisphere. Each lateral ventricle extends far into the brain, possessing three distinct "horns" (extensions):
- Anterior (Frontal) Horn: Juts sharply forward into the deep frontal lobe, ending just anterior to the interventricular foramen.
- Posterior (Occipital) Horn: Sweeps sharply backward deep into the occipital lobe.
- Inferior (Temporal) Horn: Curves sharply downward and drastically forward into the deep temporal lobe, wrapping smoothly around the thalamus.
Connection: The massive volume of CSF produced in both lateral ventricles drains exclusively into the single third ventricle through two tiny, paired bottleneck openings called the interventricular foramina of Monro.
- 3. Third Ventricle: A highly compressed, incredibly narrow, slit-like midline cavity sitting perfectly vertically between the two massive, egg-shaped thalami and hypothalami.
Connection: CSF drains from the bottom rear of the third ventricle directly into the cerebral aqueduct.
- Cerebral Aqueduct (of Sylvius): This is NOT a ventricle, but a highly critical, long, narrow pipe (measuring roughly 1-2 mm in diameter) that plunges straight through the midbrain, connecting the 3rd to the 4th ventricle. It is entirely surrounded by the periaqueductal gray.
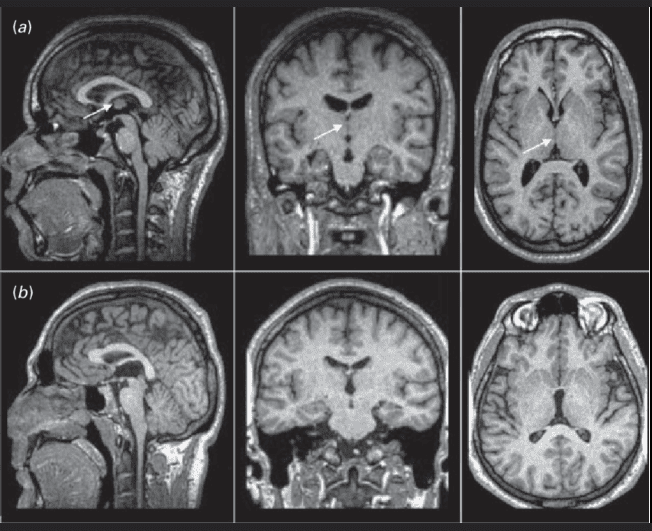
Pathology Note: Because it is a microscopic tube, it is incredibly vulnerable. A slight narrowing (aqueductal stenosis) due to congenital webs, tumors, or cellular debris will completely dam the system, causing massive upstream swelling (hydrocephalus).
- 4. Fourth Ventricle: A beautiful, diamond-shaped tent-like cavity located right where the brainstem meets the cerebellum. The anterior floor is formed by the pons and medulla (the rhomboid fossa), while the posterior roof is formed by the cerebellum.
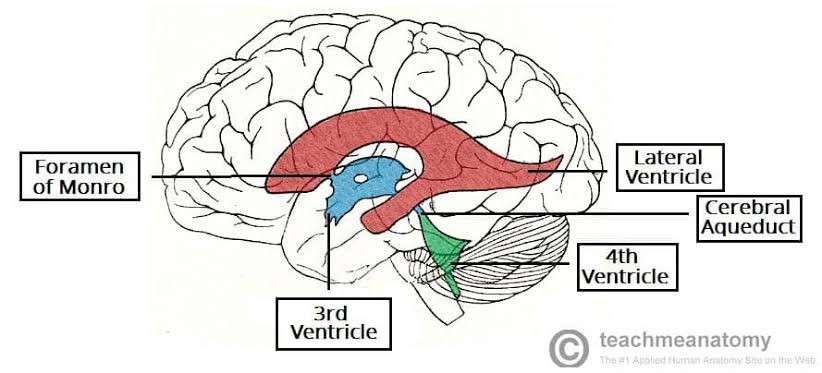
2. CSF Production, Exit Points & Flow
Production (Choroid Plexus):
CSF is not just filtered blood; it is actively secreted by the choroid plexus, a highly specialized, cauliflower-like network of dense capillary tufts completely covered by active ependymal cells. The choroid plexus is strategically located inside the ventricles. Specifically, it is found in the body and atrium of both lateral ventricles, the roof of the third ventricle, and the posterior roof of the fourth ventricle. (Note: There is NO choroid plexus in the cerebral aqueduct or the anterior/posterior horns of the lateral ventricles).
The human brain produces an astonishing 500 mL of CSF per day. However, the total physical capacity of the entire ventricular and subarachnoid system is only about 150 mL. This implies that the entire CSF volume must be constantly flushed out and fully replaced about 3 to 4 times every single day to prevent catastrophic pressure buildup.
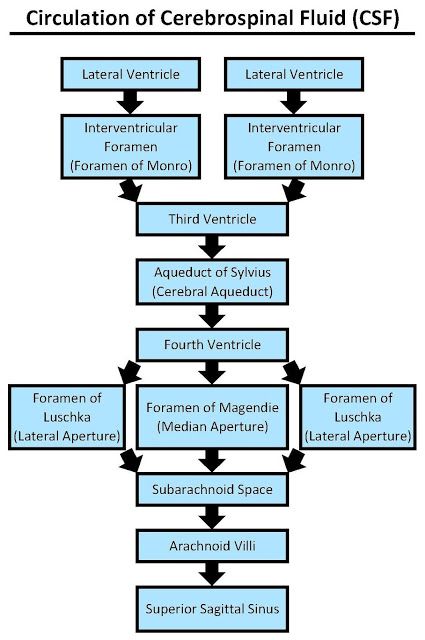
The Circulation Pathway (Step-by-Step):
- Actively produced by the heavy choroid plexus primarily in the massive Lateral Ventricles.
- Drains through the tiny Interventricular Foramina of Monro into the thin Third Ventricle (picking up more CSF here).
- Flows rapidly through the long Cerebral Aqueduct of Sylvius down into the tent-shaped Fourth Ventricle (picking up the last bit of CSF).
- To escape the brain's internal cavities and finally bathe the outside of the brain, the CSF must exit the fourth ventricle. It forces its way out through three highly specific holes (apertures) punched in the roof of the 4th ventricle:
- Lateral Apertures (of Luschka): Two paired openings, one on the far left and one on the far right, located near the cerebellopontine angle. They shoot CSF out laterally.
- Median Aperture (of Magendie): A single, large midline hole punched directly in the inferior roof. It shoots massive amounts of CSF straight back into the cisterna magna (the massive pool of CSF located perfectly between the cerebellum and the dorsal medulla).
- Once blasted out of these three holes, the CSF is now freely floating in the Subarachnoid Space, wrapping around the entire brain surface and plunging down around the spinal cord.
- Eventually, the CSF percolates up to the very top of the brain. It is finally reabsorbed back into the venous bloodstream through one-way pressure valves called arachnoid granulations (tufted projections of arachnoid mater) that punch directly into the superior sagittal venous sinus.
EXAM TIP: Mnemonic for tracing the precise Ventricular Flow:
'Lateral (2) -> Monro -> Third -> Sylvius -> Fourth -> Luschka & Magendie -> Subarachnoid.'
Mnemonic for the exit doors: 'Luschka = L = Lateral (paired, 2 openings), Magendie = M = Median (single, 1 opening).' Total = 3 apertures.
Pathophysiology Pearl
Hydrocephalus ("Water on the Brain")
Because the brain produces 500 mL of fluid daily in a rigid 150 mL box, any obstruction is a life-threatening emergency. We classify it strictly by where the dam is built:
- Non-Communicating (Obstructive) Hydrocephalus: The dam is built INSIDE the ventricular plumbing (most commonly a blockage in the tiny cerebral aqueduct, or a tumor smashing the 4th ventricle). The ventricles above the dam balloon out massively, but no fluid communicates with the subarachnoid space below.
- Communicating Hydrocephalus: The entire ventricular plumbing is perfectly open. The fluid easily exits the Luschka/Magendie doors into the subarachnoid space. The problem is a failure of reabsorption at the very end of the line. The arachnoid granulations become scarred and clogged (usually after severe meningitis or a subarachnoid hemorrhage). ALL ventricles in the brain swell symmetrically.
Infant vs. Adult: In infants, the skull bones are not yet fused, so the pressure forces the head to physically expand to massive sizes. In an adult with a fused skull, the head cannot expand; the pressure crushes the brain, causing violent headaches, projectile vomiting, papilledema (swollen optic disc), and rapid death via brain herniation unless a neurosurgeon drills a shunt into the ventricle to drain the fluid.
VI. QUICK REVIEW & EXAM CHECKLIST
This section provides a rapid-fire review of the absolute highest-yield facts and clinical correlations guaranteed to appear on anatomy practicals and medical board exams. Use it as a final, high-octane study tool before tests.
1. High-Yield Mnemonics & Memory Aids
- Basal Ganglia Core: Caudate + Putamen + Globus Pallidus = Corpus Striatum.
- Deep Cerebellar Nuclei (lateral to medial): Don't Eat Greasy Food = Dentate, Emboliform, Globose, Fastigial.
- Thalamic Geniculate Bodies: Medial = Music (auditory), Lateral = Light (visual).
- Midbrain Colliculi: Superior = Sight, Inferior = Sound.
- Medullary Tubercles: Gracile = Ground (lower body), Cuneate = Ceiling (upper body).
- Corpus Callosum Parts (front to back): Rostrum, Genu, Body, Splenium.
- Internal Capsule Limbs: Anterior Limb, Genu (carries corticobulbar to face), Posterior Limb (carries corticospinal to body).
- Cerebellar Peduncles (Traffic): Superior = Major Output, Middle = Massive Input, Inferior = Both.
2. Key Clinical Pathology Correlations
- Parkinson's Disease: Pathological death and degeneration of dopaminergic neurons specifically in the substantia nigra (the dark band easily visible on cross-section between the tegmentum and crus cerebri in the midbrain).
- Internal Capsule Stroke (Lacunar Infarct): Severe small vessel disease directly destroying the posterior limb of the internal capsule. Causes pure, dense motor or pure sensory stroke on the complete contralateral side, because all descending/ascending fibers are funneled through a tiny 1-2 mm space.
- Hydrocephalus Diagnostics: The most common, highly tested site of fatal obstruction is the tiny cerebral aqueduct (of Sylvius). Look for MRI scans showing massively dilated lateral and third ventricles, but a completely normal, unswollen fourth ventricle.
- Locked-In Syndrome: A catastrophic massive pontine lesion (almost always a basilar artery occlusion/thrombosis) that destroys all descending corticospinal and corticobulbar tracts bilaterally in the basilar pons. The patient is fully awake, fully conscious, and hears everything, but cannot move a single muscle or speak. They can communicate only via vertical eye movements (which are spared because they are controlled by the midbrain, sitting safely above the stroke).
- Hemiballismus: A devastating, localized focal lesion (stroke) destroying the tiny subthalamic nucleus in the diencephalon. Without its inhibitory "brakes," the patient suffers violent, flinging, ballistic movements on the entire contralateral side.
- Lateral Medullary (Wallenberg) Syndrome: A classic brainstem stroke (PICA occlusion) destroying the lateral, post-olivary region of the medulla. It severely damages CN IX and X, causing profound dysphagia (inability to swallow), hoarseness, vertigo, and a bizarre crossed sensory loss (loss of pain/temp on the ipsilateral face, but loss of pain/temp on the contralateral body).
- Cerebellopontine Angle (CPA) Tumors: Slow-growing, benign acoustic neuromas (schwannomas growing on CN VIII) perfectly situated at the pontomedullary junction where CN VII and VIII exit together. As the tumor swells, it crushes CN VIII (causing progressive hearing loss, severe vertigo, and tinnitus) and then crushes CN VII (causing Bell's palsy/facial weakness).
3. Exam Checklist: Can You Identify These?
Before stepping into the anatomy lab or taking a practical exam, place a mental checkmark next to each specific structure. Ensure you can confidently, rapidly locate them on a wet brain specimen, an unlabeled diagram, or an MRI cross-section:
Surface Anatomy:
■ Central sulcus, lateral fissure, parieto-occipital sulcus
■ Precentral gyrus, postcentral gyrus, hidden insular cortex
■ Frontal, parietal, temporal, occipital lobes |
White Matter:
■ Corpus callosum (rostrum, genu, body, splenium)
■ Internal capsule (anterior limb, genu, posterior limb)
■ Anterior commissure, fornix |
Deep Gray Matter:
■ Caudate nucleus (head, body, tail)
■ Putamen, globus pallidus (external and internal)
■ Amygdala, hippocampus, dentate gyrus |
Diencephalon:
■ Thalamus (anterior, medial, lateral nuclear groups)
■ Medial and lateral geniculate bodies
■ Hypothalamus (optic chiasm, infundibulum, mammillary bodies)
■ Pineal gland, habenular nuclei, subthalamic nucleus |
Midbrain:
■ Superior & inferior colliculi (corpora quadrigemina)
■ Cerebral aqueduct, periaqueductal gray
■ Red nucleus, substantia nigra
■ Crus cerebri, interpeduncular fossa, CN III/IV exits |
Pons & Cerebellum:
■ Basilar pons, basilar groove, middle cerebellar peduncles
■ CN V, CN VI/VII/VIII (pontomedullary junction)
■ Vermis, cerebellar hemispheres, anterior/posterior/flocculonodular lobes
■ Superior, middle, inferior cerebellar peduncles
■ Dentate, emboliform, globose, fastigial nuclei |
Medulla:
■ Pyramids, pyramidal decussation, olives
■ Pre-olivary sulcus (CN XII), post-olivary sulcus (CN IX, X, XI)
■ Gracile tubercle, cuneate tubercle
■ Closed medulla vs. open medulla |
Spinal Cord:
■ Conus medullaris (L1-L2), cauda equina, filum terminale
■ Cervical (C4-T1) & lumbosacral (L1-S3) enlargements
■ Dorsal/ventral/lateral horns, Anterior/lateral/posterior funiculi
■ Fasciculus gracilis, fasciculus cuneatus |
Ventricular System:
■ Lateral ventricles (anterior, posterior, inferior horns), Interventricular foramina of Monro
■ Third ventricle, cerebral aqueduct of Sylvius
■ Fourth ventricle, rhomboid fossa
■ Lateral apertures of Luschka, median aperture of Magendie, Cisterna magna, Choroid plexus locations |
Recommended Reference List
For further exhaustive study and high-resolution imaging, the following internationally recognized neuroanatomy textbooks and atlases are highly recommended to supplement this guide:
- Haines, D. E. (2018). Neuroanatomy in Clinical Context: An Atlas of Structures, Sections, Systems, and Syndromes (10th ed.). Wolters Kluwer. (Unparalleled for cross-sectional anatomy and MRIs).
- Snell, R. S. (2010). Clinical Neuroanatomy (7th ed.). Lippincott Williams & Wilkins. (Excellent for direct clinical applications and neurological deficits).
- Netter, F. H. (2019). Atlas of Human Anatomy (7th ed.). Elsevier. (The gold standard for beautiful, clear, illustrative medical art).
- Crossman, A. R., & Neary, D. (2019). Neuroanatomy: An Illustrated Colour Text (6th ed.). Elsevier. (Highly readable for rapid reviews of specific pathways).
![[IMAGE PLACEHOLDER: Cross-sectional diagram of the skull and scalp showing the 5 distinct S.C.A.L.P. layers, with emissary veins bridging the areolar tissue to the dural sinuses]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Cross-sectional-diagram-of-the-skull-and-scalp-showing-the-5-distinct-S.C.A.L.P.-layers-with-emissary-veins-bridging-the-areolar-tissue-to-the-dural-sinuses.jpg)
![[IMAGE PLACEHOLDER: Detailed anterior and lateral views of the face illustrating the complex network of facial muscles interacting around the eyes, nose, and mouth]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Detailed-anterior-and-lateral-views-of-the-face-illustrating-the-complex-network-of-facial-muscles-interacting-around-the-eyes-nose-and-mouth-1-1.png)
![[IMAGE PLACEHOLDER: Lateral view of the head mapping the sensory dermatomes of V1, V2, and V3 (Trigeminal), alongside the cervical plexus territories (C2, C3) on the scalp and neck]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-head-mapping-the-sensory-dermatomes-of-V1-V2-and-V3.jpg)
![[IMAGE PLACEHOLDER: Diagram of the Facial Nerve exiting the stylomastoid foramen, branching out through the translucent parotid gland into its 5 terminal branches on the face]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Diagram-of-the-Facial-Nerve-exiting-the-stylomastoid-foramen-branching-out-through-the-translucent-parotid-gland-into-its-5-terminal-branches-on-the-face.png)
![[IMAGE PLACEHOLDER: Lateral vascular map of the head and neck, showing the External Carotid Artery branching into the tortuous Facial Artery and ascending Superficial Temporal Artery, alongside the major venous drainage routes]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-vascular-map-of-the-head-and-neck-showing-the-External-Carotid-Artery-branching-into-the-tortuous-Facial-Artery-1-1.png)
![[IMAGE PLACEHOLDER: Anterior view of the neck illustrating the Sternocleidomastoid muscle dividing the neck into the Anterior and Posterior Triangles, mapped with their specific subdivisions]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-view-of-the-neck-illustrating-the-Sternocleidomastoid-muscle-dividing-the-neck-into-the-Anterior-and-Posterior-Triangles-mapped-with-their-specific-subdivisions-1.jpg)
![[IMAGE PLACEHOLDER: Lateral view of the head and upper torso, highlighting the neck region in yellow. Shows the Superior nuchal line, Mastoid process, Mandible, Vertebra C7, Clavicle, Manubrium of sternum, and Acromion]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-head-and-upper-torso.webp)
![[IMAGE PLACEHOLDER: Cross-section of the neck showing the fascial layers and compartments. Anterior shows the Pretracheal fascia enclosing the Visceral compartment. Lateral shows the Carotid sheath enclosing the Vascular compartments. Posterior shows the Prevertebral fascia enclosing the Vertebral compartment. The Investing fascia surrounds the entire outer layer.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Cross-section-of-the-neck-showing-the-fascial-layers-and-compartments.webp)
![[IMAGE PLACEHOLDER: Lateral view of the neck illustrating the Anterior Triangle (outlined in green) and Posterior Triangle (outlined in blue). Shows the SCM muscle dividing the two, the mandible forming the superior border, and the clavicle forming the base.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-neck-illustrating-the-Anterior-Triangle.webp)
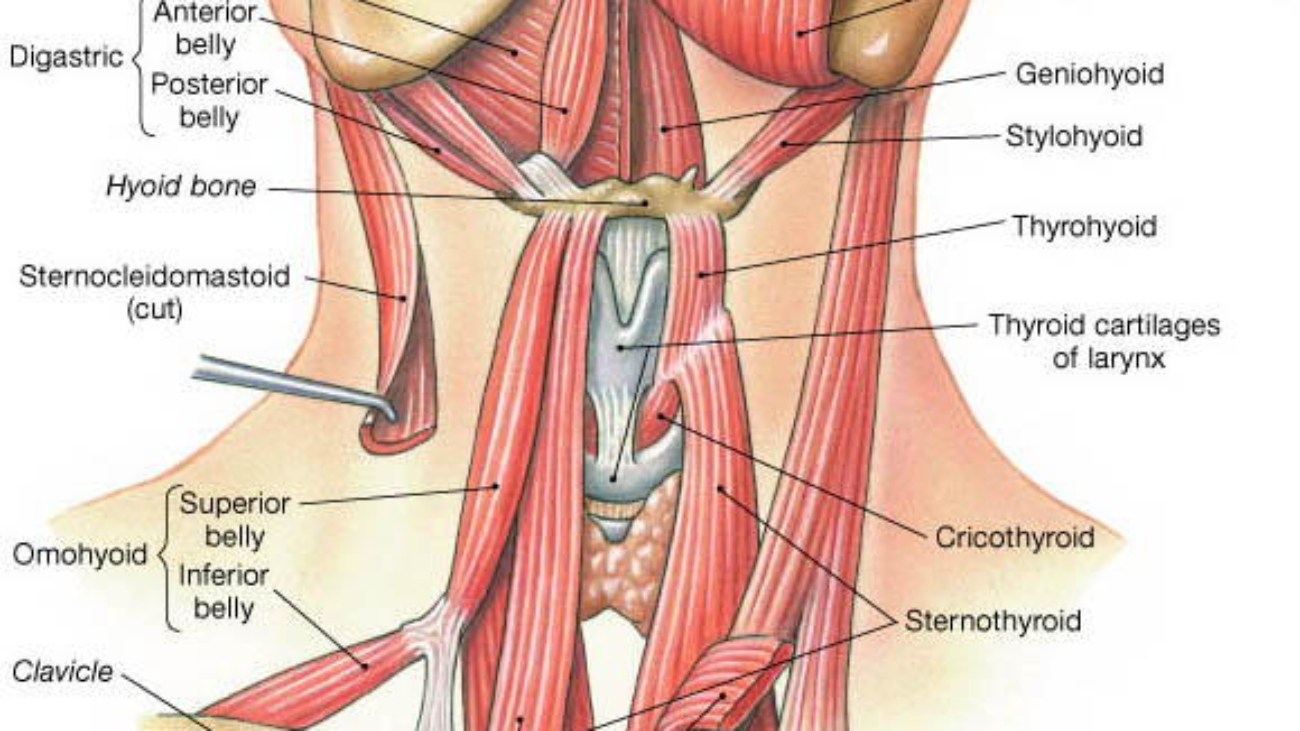
![[IMAGE PLACEHOLDER: Detailed diagram of the subdivided Anterior Triangle. Clearly labels the Submandibular, Submental, Muscular, and Carotid triangles defined by the digastric and omohyoid muscles.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Detailed-diagram-of-the-subdivided-Anterior-Triangle.-Clearly-labels-the-Submandibular-Submental-Muscular-and-Carotid-triangles-defined-by-the-digastric-and-omohyoid-muscles.webp)
![[IMAGE PLACEHOLDER: Two views of the Suprahyoid muscles. View A (lateral) showing Stylohyoid, Digastric (anterior and posterior bellies), and Mylohyoid. View B (inferior/anterior) showing Geniohyoid and Mylohyoid forming the floor of the mouth.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Two-views-of-the-Suprahyoid-muscles.jpeg)
![[IMAGE PLACEHOLDER: Anterior view of the neck exposing the Infrahyoid (Strap) muscles. Shows the Hyoid bone, Thyroid cartilage, Omohyoid, Sternohyoid, Thyrohyoid, and Sternothyroid muscles, alongside the IJV and Carotid artery.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-view-of-the-neck-exposing-the-Infrahyoid.webp)
![[IMAGE PLACEHOLDER: Diagram showing the origin of the common carotid arteries. The right common carotid branching from the brachiocephalic trunk, and the left common carotid branching directly from the aortic arch.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/carotid-artery.jpg)
![[IMAGE PLACEHOLDER: Medial view of the right carotid artery bifurcation. Clearly circles the swollen Carotid Sinus and points to the small, nodular Carotid Body nestled in the crotch of the bifurcation. Shows Glossopharyngeal nerve branches attaching to them.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Medial-view-of-the-right-carotid-artery-bifurcation.webp)
![[IMAGE PLACEHOLDER: Two illustrations of neck nerves. Left shows the Transverse cervical nerve providing sensory coverage over the SCM. Right shows the intricate loop of the Ansa Cervicalis overlying the IJV, sending motor branches into the strap muscles.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/ansa-cervicalis-1-2-1.gif)
![[IMAGE PLACEHOLDER: Collage of four diagrams highlighting specific cranial nerves. Shows the Glossopharyngeal [IX], Vagus [X], Accessory [XI], and Hypoglossal [XII] nerves routing through the neck musculature and vasculature.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/neck-nerves.jpg)
![[IMAGE PLACEHOLDER: Anterior and cross-sectional views of the Thyroid gland. Shows the two lateral lobes and central isthmus overlying the trachea. Cross-section emphasizes the Pretracheal fascia wrapping the thyroid, trachea, and esophagus together.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-and-cross-sectional-views-of-the-Thyroid-gland.webp)
![[IMAGE PLACEHOLDER: Detailed arterial mapping of the Thyroid Gland. Shows the Superior thyroid artery descending from the External Carotid, and the Inferior thyroid artery ascending from the Thyrocervical trunk of the Subclavian artery.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/vascular-thyroid.webp)
![[IMAGE PLACEHOLDER: Posterior view of the Trachea and Thyroid showing the Left and Right Recurrent Laryngeal Nerves looping under major vessels and ascending tightly in the groove between the trachea and esophagus, running directly behind the thyroid lobes.]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Posterior-view-of-the-Trachea-and-Thyroid-showing-the-Left-and-Right-Recurrent-Laryngeal-Nerves.webp)
![[IMAGE PLACEHOLDER: Full body skeleton highlighting the Axial Skeleton in blue and Appendicular Skeleton in pink]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Full-body-skeleton-highlighting-the-Axial-Skeleton-in-blue-and-Appendicular-Skeleton-in-pink.webp)
![[IMAGE PLACEHOLDER: Lateral view of the skull showcasing the Cranial vs. Facial divisions, highlighting major sutures and bone regions]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-skull-showcasing-the-Cranial-vs.-Facial-divisions-highlighting-major-sutures-and-bone-regions.webp")
![[IMAGE PLACEHOLDER: Anterior view of the Frontal Bone detailing the Squama, Supraorbital Foramen, and Superciliary Arches]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-view-of-the-Frontal-Bone-detailing-the-Squama-Supraorbital-Foramen-and-Superciliary-Arche.webp)
![[IMAGE PLACEHOLDER: Lateral view of Parietal bones highlighting the Sagittal, Coronal, Lambdoid, and Squamous sutures]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-Parietal-bones-highlighting-the-Sagittal-Coronal-Lambdoid-and-Squamous-sutures-1.jpg)
![[IMAGE PLACEHOLDER: Inferior/Posterior view of the Occipital bone showing the Foramen Magnum, Occipital Condyles, and Nuchal Lines]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Inferior-Posterior-view-of-the-Occipital-bone-showing-the-Foramen-Magnum-Occipital-Condyles-and-Nuchal-Lines.jpg)
![[IMAGE PLACEHOLDER: Superior and Anterior views of the Sphenoid Bone resembling a bat, highlighting the Sella Turcica, Greater/Lesser Wings, and Pterygoid Processes]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Superior-and-Anterior-views-of-the-Sphenoid-Bone-resembling-a-bat-highlighting-the-Sella-Turcica-Greater-Lesser-Wings-and-Pterygoid-Processes.webp)
![[IMAGE PLACEHOLDER: Lateral view of the Temporal Bone showing the Squamous portion, Mastoid process, Styloid process, and Zygomatic process]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-Temporal-Bone-showing-the-Squamous-portion-Mastoid-process-Styloid-process-and-Zygomatic-process.jpg)
![[IMAGE PLACEHOLDER: Anterior/Superior view of the Ethmoid Bone showing the Crista Galli, Cribriform Plate, Perpendicular Plate, and Conchae]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-Superior-view-of-the-Ethmoid-Bone-showing-the-Crista-Galli-Cribriform-Plate-Perpendicular-Plate-and-Conchae-1.jpg)
![[IMAGE PLACEHOLDER: Anterior view of the Maxilla showing the Alveolar process, Palatine process, and Infraorbital foramen]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-view-of-the-Maxilla-showing-the-Alveolar-process-Palatine-process-and-Infraorbital-foramen.png)
![[IMAGE PLACEHOLDER: Lateral view of the Mandible showing the Body, Ramus, Angle, Condylar process, Coronoid process, and Mental foramen]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-view-of-the-Mandible-showing-the-Body-Ramus-Angle-Condylar-process-Coronoid-process-and-Mental-foramen.jpg)
![[IMAGE PLACEHOLDER: Anterior skull view isolating the Vomer and Inferior Nasal Conchae inside the nasal cavity]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Anterior-skull-view-isolating-the-Vomer-and-Inferior-Nasal-Conchae-inside-the-nasal-cavity-3.jpg)
![[IMAGE PLACEHOLDER: Lateral and Posterior views of the full Vertebral Column, color-coded to show Cervical, Thoracic, Lumbar, Sacral, and Coccyx regions]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Lateral-and-Posterior-views-of-the-full-Vertebral-Column-color-coded-to-show-Cervical-Thoracic-Lumbar-Sacral-and-Coccyx-regions.jpg)
![[IMAGE PLACEHOLDER: Visual comparison of the Atlas (C1) ring structure, the Axis (C2) highlighting the Dens, and the articulation complex showing how C1 pivots on C2]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Bony-Landmarks-of-the-Atlas-and-Axis.png)
![[IMAGE PLACEHOLDER: Superior view of a Typical Cervical Vertebra showing the small Body, large triangular Vertebral Foramen, Bifid Spinous Process, and Transverse Foramina]](https://doctorsrevisionuganda.com/wp-content/uploads/2026/06/Superior-view-of-a-Typical-Cervical-Vertebra-showing-the-small-Body-large-triangular-Vertebral-Foramen-Bifid-Spinous-Process-and-Transverse-Foramina.webp)