Biochemistry: Gluconeogenesis Exam
Test your knowledge with these 30 questions.
Gluconeogenesis Exam
Question 1/30
Exam Complete!
Here are your results, .
Your Score
28/30
93%
Test your knowledge with these 30 questions.
Question 1/30
Here are your results, .
Your Score
28/30
93%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
Test your knowledge with these 30 questions.
Question 1/30
Here are your results, .
Your Score
27/30
90%
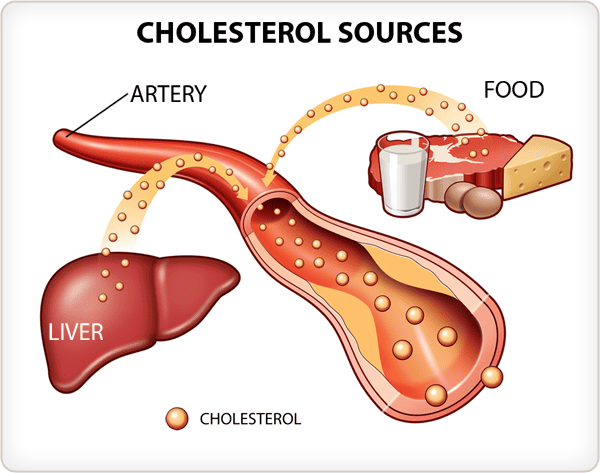
Cholesterol often gets a bad rap due to its association with heart disease, but it's crucial to understand that it is an essential molecule for life. Our bodies need cholesterol to function properly. The problem arises when its levels are imbalanced or when it's handled improperly within the body.
Cholesterol is a lipid belonging to the steroid family. Its unique amphipathic structure (a polar hydroxyl group and a nonpolar steroid ring system and hydrocarbon tail) allows it to insert into cell membranes, giving it critical structural and signaling roles.
The body acquires cholesterol from two main sources:
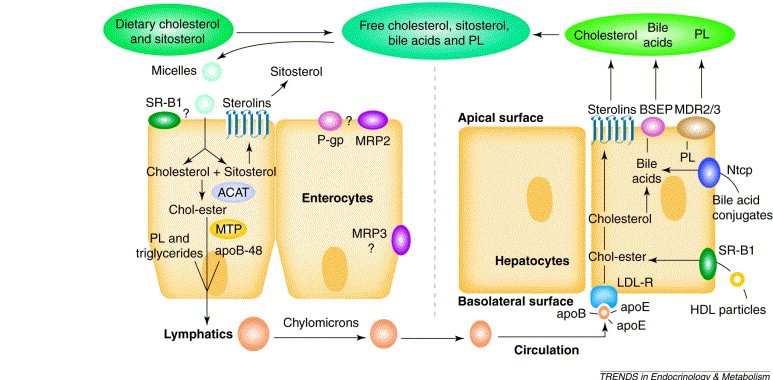
The process of dietary cholesterol absorption primarily occurs in the small intestine:
So, to summarize, cholesterol is a vital molecule for cell structure, hormones, bile acids, and Vitamin D. We get it from both our diet and internal synthesis. Dietary cholesterol is absorbed in the small intestine via NPC1L1, processed, and then packaged into chylomicrons for transport.
Cholesterol is an indispensable molecule, and while we obtain some from our diet, the human body possesses the remarkable ability to synthesize nearly all the cholesterol it requires through a complex process known as de novo synthesis. This internal production ensures a constant supply for vital cellular functions.
While virtually all nucleated cells can synthesize cholesterol, certain tissues are particularly active:
The enzymatic machinery is distributed between two key cellular compartments:
The synthesis of cholesterol is an energetically demanding process:
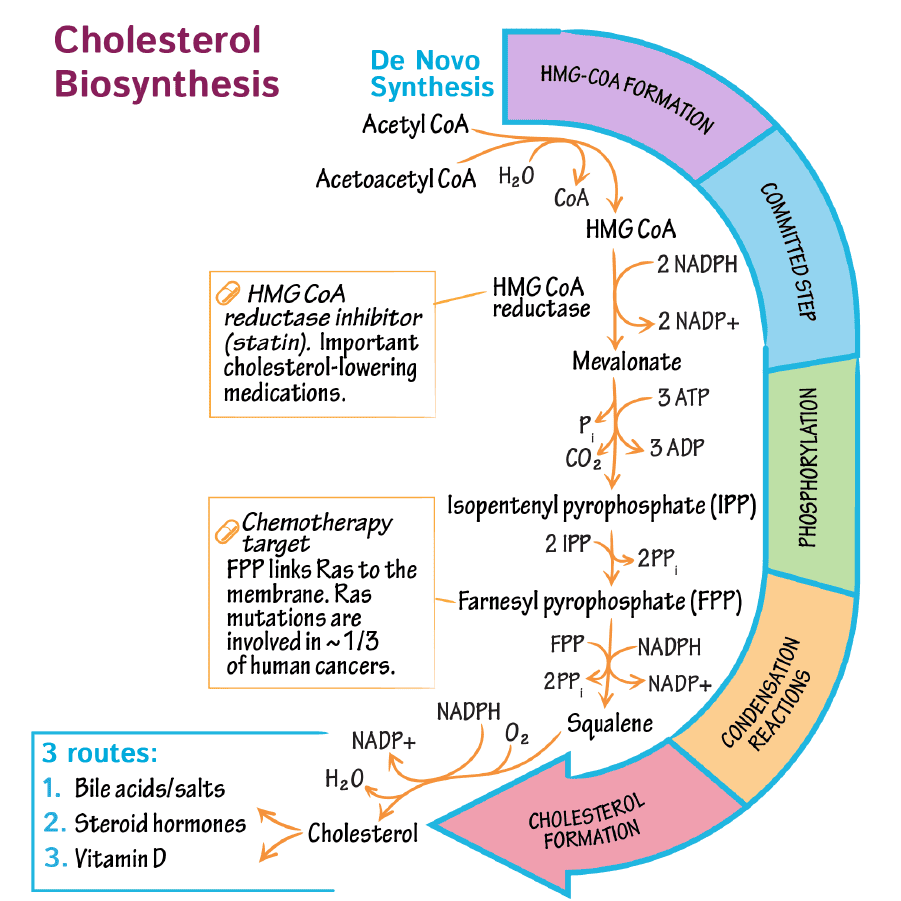
The complex pathway can be delineated into five principal stages:
The synthesis initiates with the condensation of Acetyl-CoA units:
It is crucial to note the distinction from ketone body synthesis: the cytosolic HMG-CoA synthase produces HMG-CoA for cholesterol synthesis, while the mitochondrial HMG-CoA synthase participates in ketogenesis. This segregation ensures the pathways operate independently.
This stage represents the rate-limiting and committed step in cholesterol biosynthesis:
Mevalonate is subsequently processed to generate activated 5-carbon units:
The activated 5-carbon isoprenoid units are progressively linked:
The linear squalene molecule undergoes cyclization and a series of modifications:
These precise modifications culminate in the formation of cholesterol.
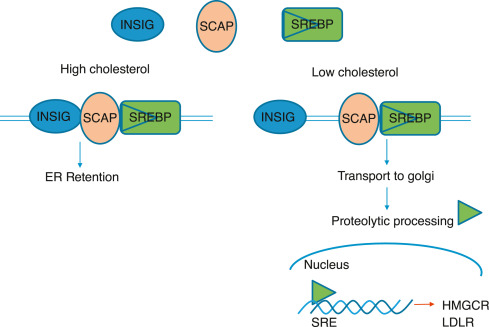
The synthesis of cholesterol is a highly regulated process. The primary point of control is the enzyme HMG-CoA reductase, the rate-limiting step in the pathway. Regulation occurs through several sophisticated mechanisms:
High concentrations of cholesterol also exert an inhibitory effect on the translation of HMG-CoA reductase mRNA, directly reducing the quantity of enzyme synthesized.
High sterol levels induce a conformational change in the reductase enzyme, making it more susceptible to ubiquitination and subsequent degradation by the proteasome. This shortens the enzyme's lifespan, leading to a quick reduction in its activity.
HMG-CoA reductase exists in two interconvertible forms:
Phosphorylation is primarily mediated by AMP-activated protein kinase (AMPK), which is activated when cellular ATP is low. By phosphorylating and inactivating HMG-CoA reductase, AMPK conserves cellular energy.
Hormonal Influence:
Bile acids, which are derivatives of cholesterol, can also contribute to feedback regulation by inhibiting HMG-CoA reductase activity.
While not a direct regulatory mechanism for synthesis, the major players in cholesterol transport are intrinsically linked to overall cholesterol homeostasis:
Bile acids are a family of steroid acids that represent the major catabolic products of cholesterol in the body. Their primary physiological function is to facilitate the digestion and absorption of dietary fats and fat-soluble vitamins in the small intestine. They also play a crucial role in cholesterol homeostasis by being the principal route for cholesterol excretion.
Bile is a complex, watery, yellowish-green fluid produced by the liver. It consists of a watery mixture of organic and inorganic compounds.
The quantitatively most important organic components of bile are phosphatidylcholine (lecithin) and conjugated bile salts.
Bile can either pass directly from the liver into the duodenum (the first part of the small intestine) via the common bile duct, or it can be stored and concentrated in the gallbladder when not immediately needed for digestion.
The synthesis of bile acids, known as cholic acid and chenodeoxycholic acid, occurs exclusively in the liver. This multi-step pathway converts the hydrophobic cholesterol molecule into more polar, amphipathic bile acids, making them water-soluble.
The synthesis pathway involves the insertion of hydroxyl groups at specific positions on the steroid structure of cholesterol. The hydrocarbon chain is also shortened by three carbons.
The first and rate-limiting step in bile acid synthesis is the introduction of a hydroxyl group at carbon 7 of cholesterol, forming 7α-hydroxycholesterol.
This reaction is catalyzed by the enzyme cholesterol 7α-hydroxylase (CYP7A1).
CYP7A1 is a cytochrome P450 enzyme, requiring molecular oxygen (O₂) and NADPH.
Regulation: The activity of CYP7A1 is highly regulated. It is inhibited by bile acids (a feedback mechanism) and induced by cholesterol (when cholesterol levels are high). This ensures that bile acid synthesis is responsive to both bile acid demand and cholesterol availability.
Following the initial hydroxylation, 7α-hydroxycholesterol undergoes a series of additional modifications. These steps involve:
These reactions ultimately lead to the formation of the two primary bile acids:
To significantly improve their ability to emulsify fat and enhance their water solubility, primary bile acids are further modified in the liver through a process called conjugation. They are joined with either the amino acid glycine or taurine.
The carboxyl group (–COOH) at the end of the bile acid side chain forms an amide bond with the amino group (–NH₂) of glycine or taurine.
This reaction is catalyzed by bile acid-CoA ligase (which activates the bile acid by forming a CoA thioester) and bile acid-CoA:amino acid N-acyltransferase.
This generates the conjugated bile acids:
These conjugated forms are all necessary to give bile its essential function in fat digestion.
At physiological pH, these conjugated bile acids exist as anions (negatively charged) due to the low pKa of their conjugates. Therefore, they are referred to as bile salts (e.g., taurocholate, glycocholate). The term "bile salts" specifically refers to these ionized forms.
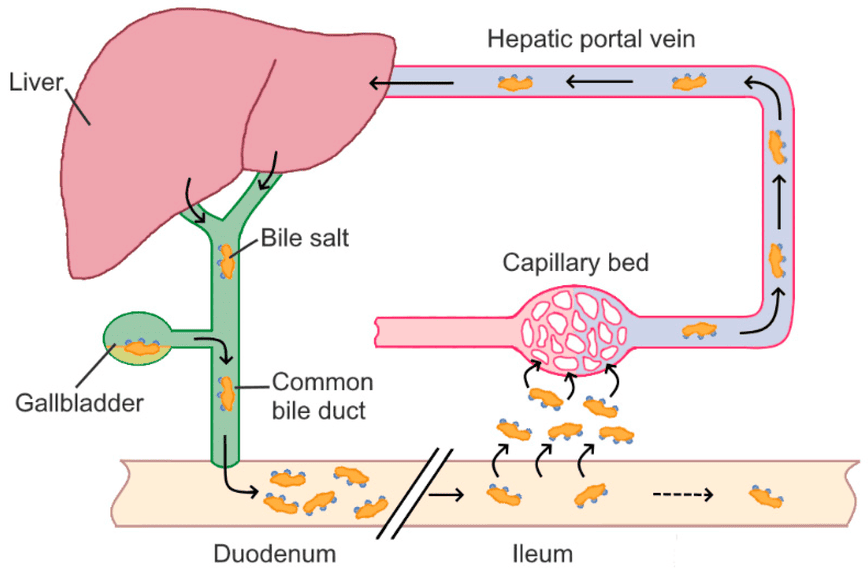
Bile salts are essential for fat digestion, but the body has a highly efficient system to conserve them rather than synthesizing new ones for every meal. This system is called the enterohepatic circulation.
Synthesized and conjugated bile salts are secreted from the liver, stored in the gallbladder, and released into the duodenum after a fatty meal.
In the duodenum and jejunum, bile salts emulsify dietary fats and form mixed micelles.
A remarkable 95% of bile salts are reabsorbed in the ileum (the final part of the small intestine). This reabsorption occurs via a specialized, active transport system known as the apical sodium-dependent bile acid transporter (ASBT) in the ileal enterocytes. Some passive reabsorption of unconjugated bile acids can also occur in the jejunum and colon.
Once reabsorbed, bile salts enter the portal venous blood and are transported back to the liver, mostly bound to albumin.
The liver efficiently extracts the bile salts from the portal blood via specific transporters.
The liver then re-secretes these reabsorbed bile salts into the bile, completing the circulation. This cycle can occur 4-12 times a day.
Not all bile acids are reabsorbed directly. Bacterial action in the gut leads to the formation of secondary bile acids.
As bile salts travel through the colon, intestinal bacteria can deconjugate them, removing glycine or taurine.
These free primary bile acids can then be further metabolized by gut bacteria, specifically undergoing 7α-dehydroxylation. This results in the formation of secondary bile acids:
Most secondary bile acids are also reabsorbed and return to the liver. In the liver, deoxycholic acid can be re-conjugated. Lithocholic acid, which is less soluble, is often sulfonated before being secreted back into bile, which aids in its excretion.
The excretion of cholesterol from the body primarily occurs via two main routes:
Cholesterol is not merely a structural component of cell membranes or a precursor for bile acids; it is also the obligate precursor for all steroid hormones. These powerful signaling molecules regulate a vast array of physiological processes, including metabolism, inflammation, immune responses, salt and water balance, sexual development, and reproduction.
The synthesis of all steroid hormones follows a common, fundamental pathway that begins with cholesterol. This process primarily occurs in the mitochondria and endoplasmic reticulum of steroidogenic tissues.
While virtually all cells contain cholesterol, steroid hormone synthesis is restricted to specialized endocrine tissues, including:
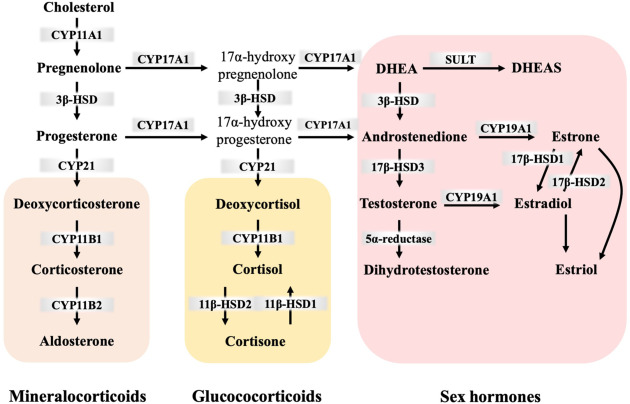
From pregnenolone, the pathway diverges. The specific hormones produced depend on the enzymatic machinery present in the particular tissue.
The synthesis is tightly regulated by the hypothalamic-pituitary-adrenal/gonadal axes.
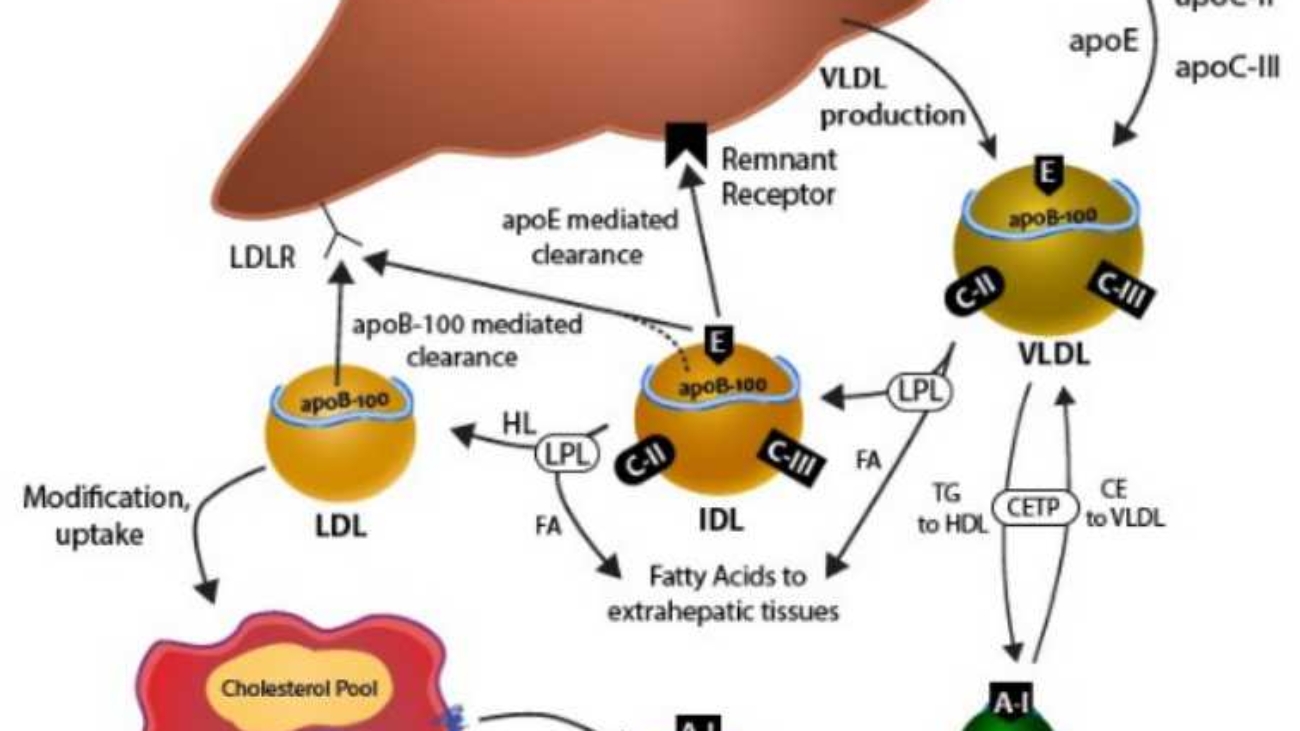
Cholesterol, being a lipid, is largely insoluble in the aqueous environment of blood plasma. To be efficiently transported between tissues for synthesis, utilization, and excretion, cholesterol (along with other lipids like triglycerides and phospholipids) is packaged into complex particles called lipoproteins. These molecular transporters have a hydrophilic exterior and a hydrophobic core, allowing them to carry lipids through the bloodstream.
Cholesteryl ester in the diet is hydrolyzed to cholesterol, which is then absorbed by the intestine together with dietary unesterified cholesterol and other lipids. It is then incorporated into chylomicrons.
Ninety-five percent of the chylomicron cholesterol is delivered to the liver in chylomicron remnants. Most of the cholesterol secreted by the liver in VLDL is retained during the formation of IDL and ultimately LDL, which is taken up by the LDL receptor in liver and extrahepatic tissues.
All lipoproteins share a common structural organization:
Lipoproteins are classified based on their density (more lipid = less dense). From largest/least dense to smallest/most dense, the main classes are:
Maintaining cholesterol homeostasis is critical. The body employs an intricate network of regulatory mechanisms, with the primary point of control being the enzyme HMG-CoA reductase.
The tight regulation is vital because both insufficient (hypocholesterolemia) and excessive (hypercholesterolemia) cholesterol levels are detrimental. Excess cholesterol, particularly carried by LDL, can lead to its deposition in arterial walls, causing atherosclerosis.
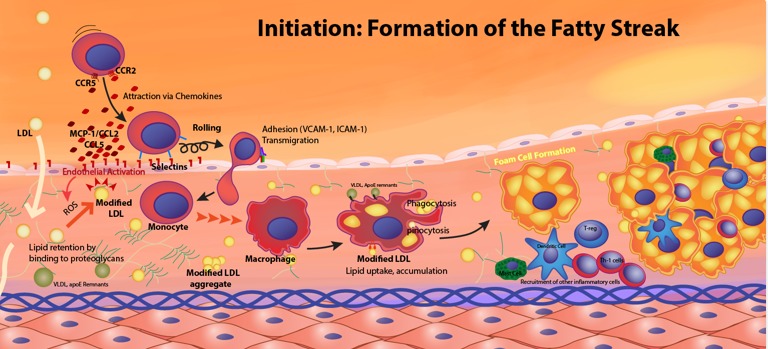
Atherosclerosis is a chronic inflammatory disease characterized by the buildup of fatty plaques within the arterial walls, leading to hardening and narrowing of the arteries.
The development of atherosclerotic plaques is a multi-stage process:
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
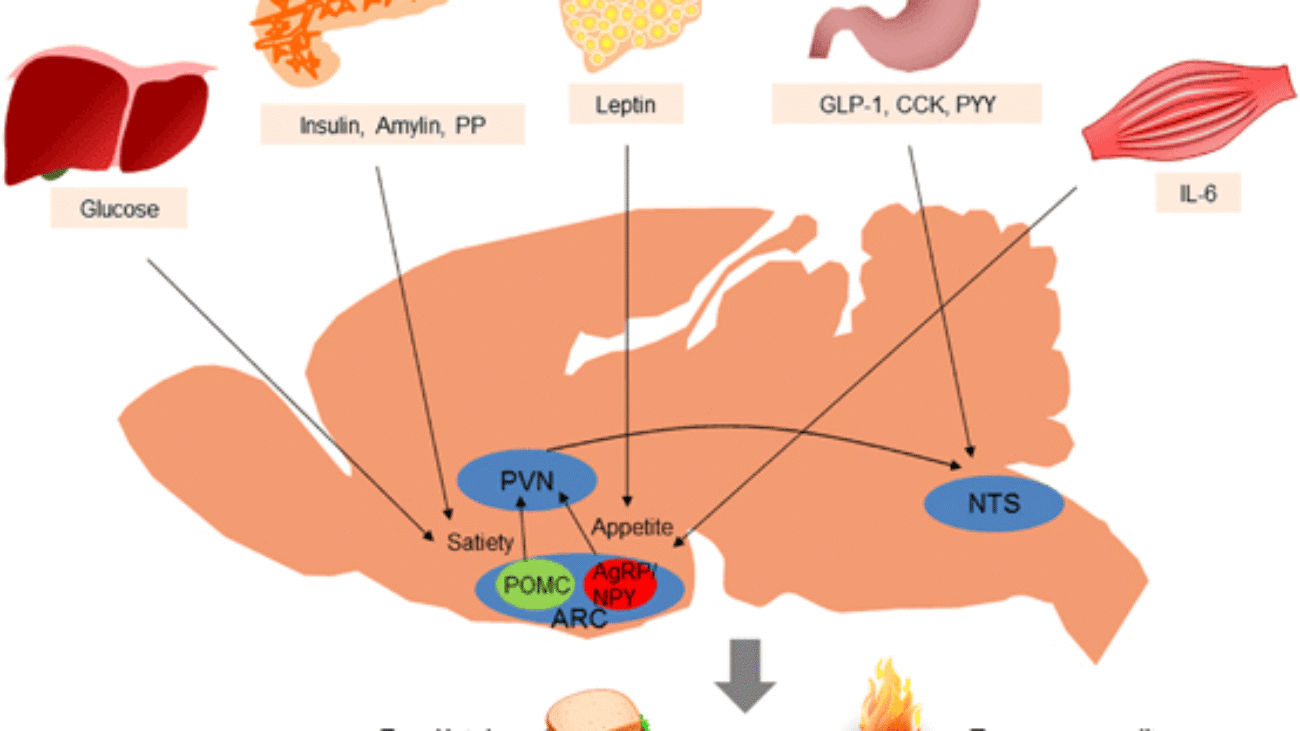
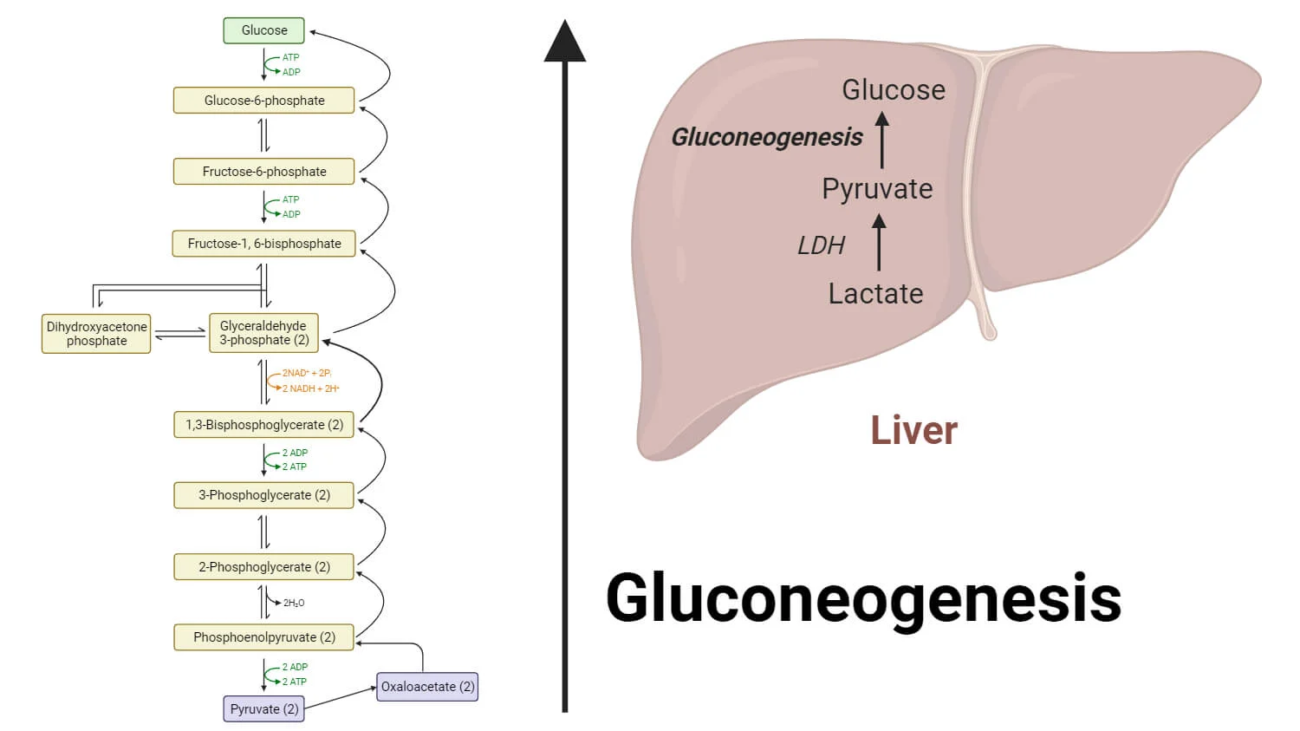
Fuel Homeostasis refers to the dynamic equilibrium and finely tuned regulation of energy substrates (glucose, fatty acids, ketone bodies, amino acids) in the body. Its primary goal is to ensure a continuous and adequate supply of fuel to all tissues, particularly the brain, under varying physiological conditions.
It is crucial for survival, allowing the body to adapt to fluctuations in nutrient availability and energy demand. Disruptions lead to metabolic diseases like diabetes, obesity, and metabolic syndrome.
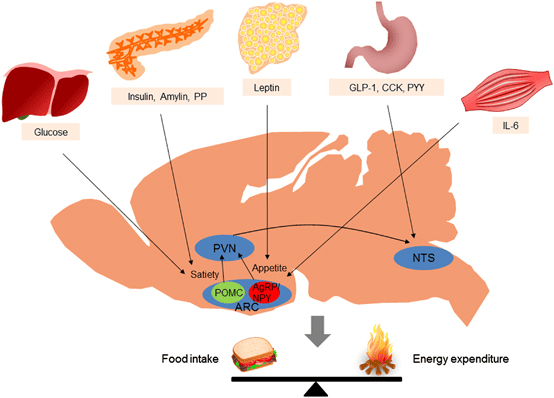
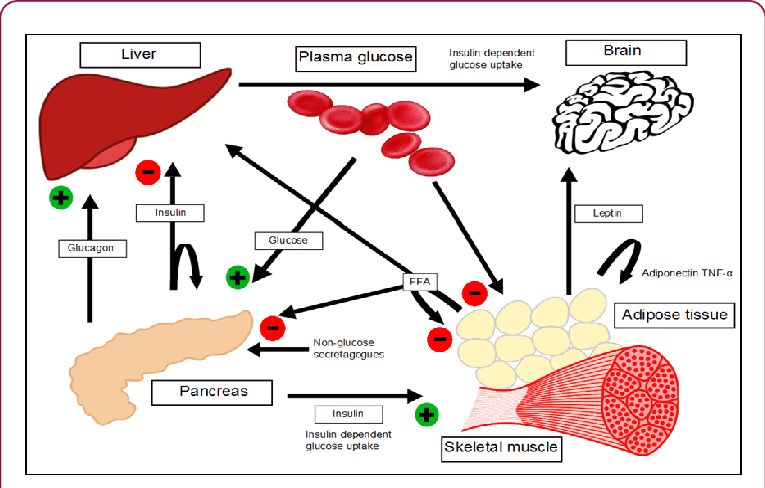
The human body is a highly integrated system where different organs specialize in fuel storage, production, and utilization.
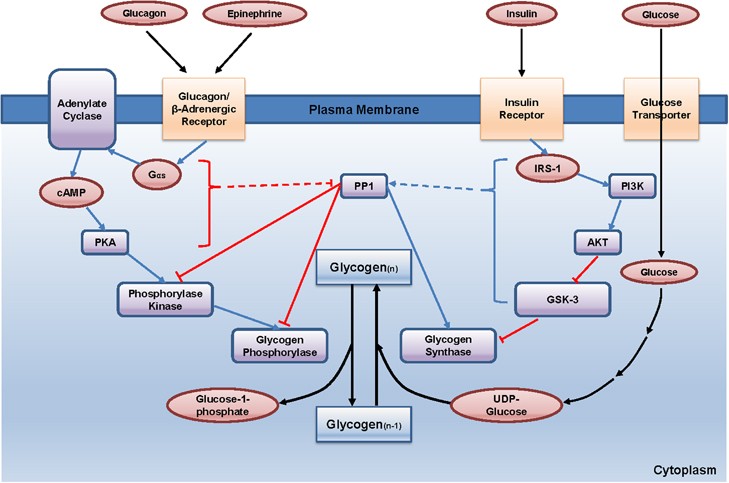
These hormones act synergistically and antagonistically to maintain metabolic balance.
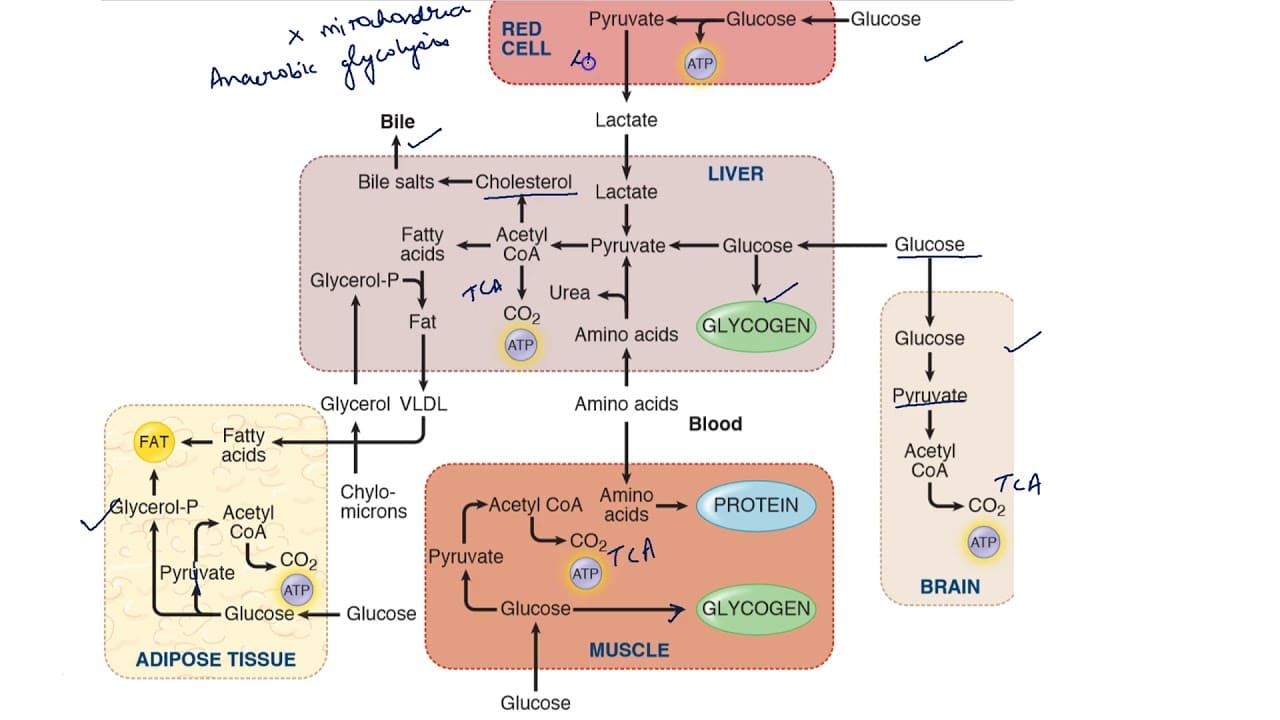
The fed state is characterized by nutrient absorption from the gastrointestinal tract, leading to elevated levels of glucose, amino acids, and triacylglycerols in the blood. The body's primary response is to store these excess nutrients and utilize glucose as the main fuel.
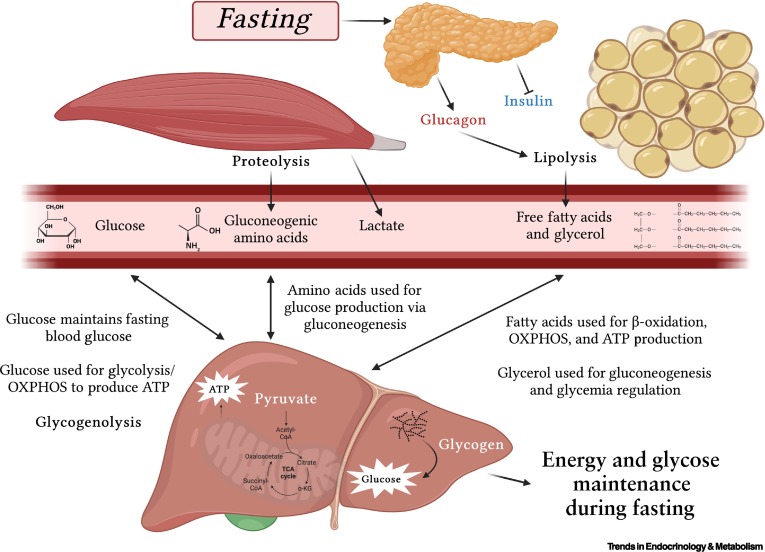
The fasting state is characterized by the absence of nutrient intake. The body must now shift from storing fuels to mobilizing its endogenous reserves to maintain a steady supply of energy, especially for the brain. This transition is orchestrated by a low insulin:glucagon ratio.
The primary goal is to maintain blood glucose levels for the brain and other glucose-dependent tissues.
To conserve glucose for the brain, other tissues switch their fuel preference to fatty acids and ketone bodies.
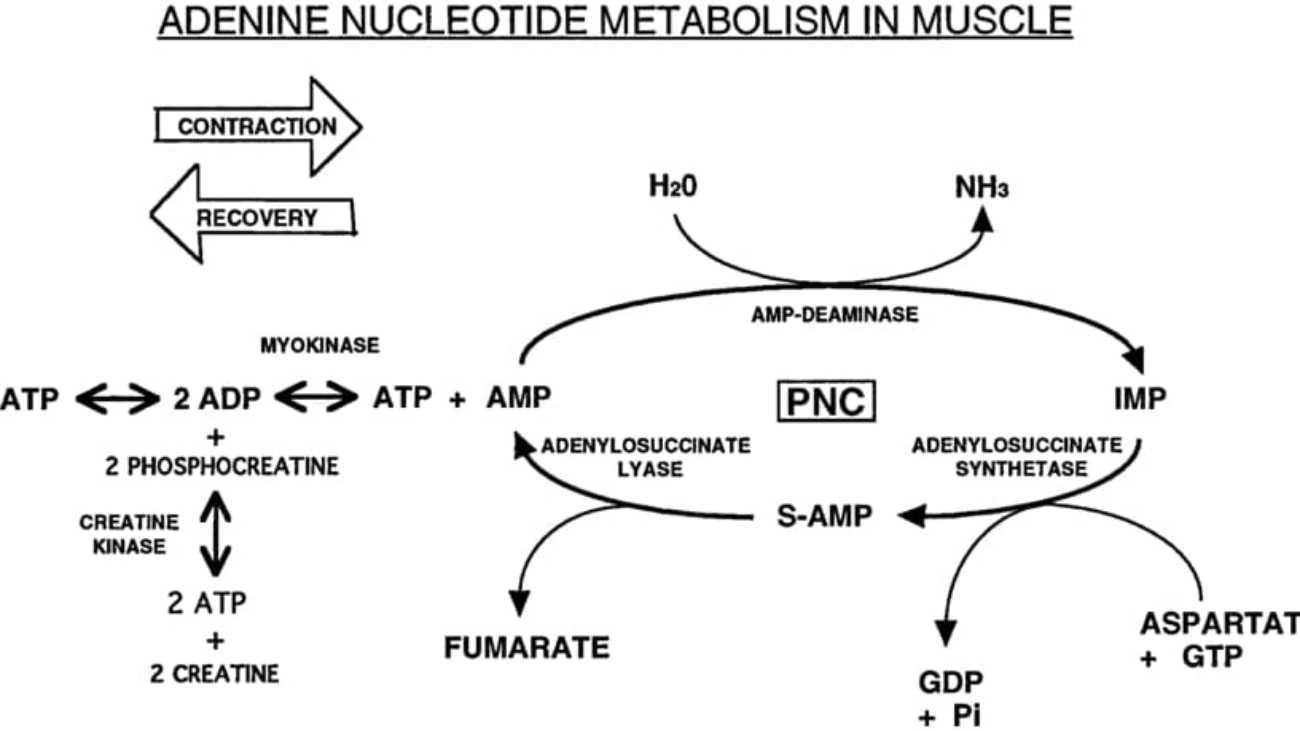
The amino groups removed from amino acids are converted to ammonia, which is detoxified in the liver via the urea cycle, producing urea for excretion. The rate of the urea cycle increases during fasting.
In summary, the early fasting state is a period of catabolism driven by a low insulin:glucagon ratio. The body prioritizes maintaining blood glucose through glycogenolysis and gluconeogenesis, while other tissues shift to fatty acid oxidation. Ketone body production begins to ramp up, setting the stage for their increased utilization in prolonged starvation.
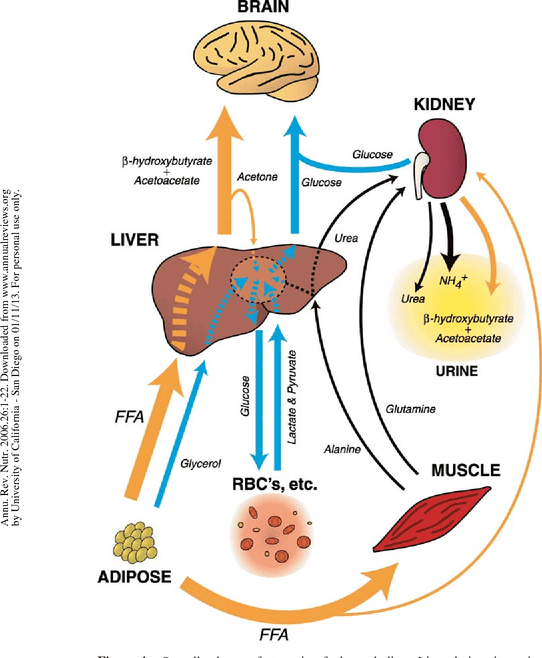
The starved state represents an extended period of nutrient deprivation, pushing the body's metabolic adaptations to their limits. The primary goals shift to:
By the time the starved state is reached (typically after 24-48 hours), liver glycogen stores are almost completely depleted. The body can no longer rely on glycogenolysis.
Lipolysis in adipose tissue continues at a very high rate, providing a continuous supply of fatty acids (for fuel) and glycerol (for gluconeogenesis). Fat stores are the largest energy reserve.
The liver's production of ketone bodies reaches its peak. The high influx of fatty acids, coupled with the low insulin state, promotes maximal β-oxidation and subsequent conversion of Acetyl-CoA into acetoacetate and β-hydroxybutyrate. Blood ketone body levels rise to very high concentrations, serving as the primary fuel for the brain, heart, and skeletal muscle.
After an initial period of high protein catabolism, the body adapts to significantly reduce muscle protein breakdown. This is directly linked to the brain's increased use of ketone bodies, as less glucose needs to be synthesized from amino acids. This adaptation is critical for long-term survival.
As amino acid catabolism decreases, the amount of nitrogen released also decreases. Consequently, the liver's production of urea via the urea cycle significantly declines. This is reflected in a reduced excretion of urea in the urine, signifying the shift to protein-sparing metabolism.
Summary of the Starved State: The starved state is characterized by extreme adaptations aimed at survival. The body shifts almost entirely to fat and ketone body metabolism to preserve its vital protein reserves. The brain becomes a major consumer of ketone bodies, dramatically reducing its glucose requirement and allowing for a significant reduction in the breakdown of muscle protein. This allows individuals to survive for extended periods without food.
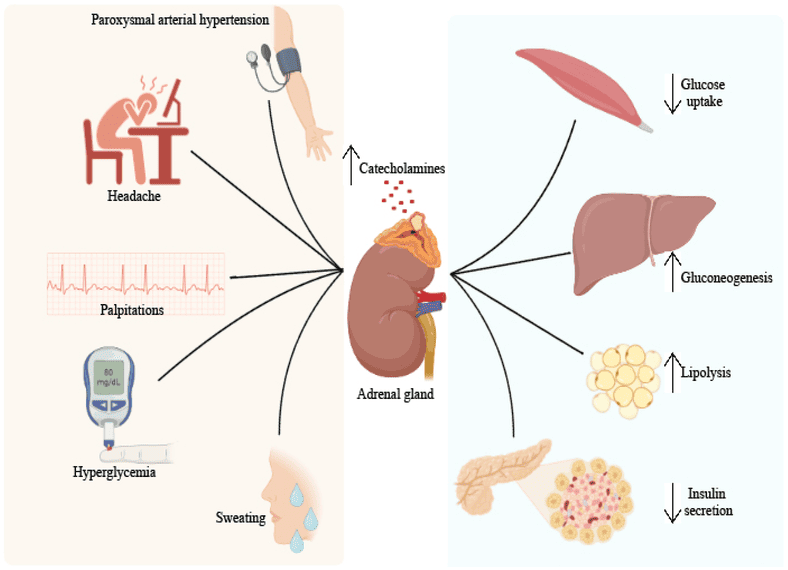
Diabetes Mellitus (DM) is a group of metabolic diseases characterized by hyperglycemia (high blood glucose) resulting from defects in insulin secretion, insulin action, or both. This chronic hyperglycemia is associated with long-term damage and failure of various organs.
The core problem is a breakdown in the body's ability to regulate glucose, leading to a state that inappropriately resembles a constant "fasted" or even "starved" state in some tissues, despite abundant glucose in the blood.
This leads to a profound metabolic crisis, an exaggerated fasted state, if untreated.
The absence of insulin inhibits protein synthesis and promotes muscle protein breakdown. The released amino acids contribute to hepatic gluconeogenesis, exacerbating hyperglycemia and leading to significant weight loss.
Insulin resistance leads to increased lipolysis, increased VLDL production, low HDL cholesterol, and the formation of small, dense LDL particles, increasing cardiovascular disease risk.
Patients with T2DM usually produce some insulin, which is often enough to suppress massive ketogenesis. A more common acute complication is Hyperosmolar Hyperglycemic State (HHS), characterized by extreme hyperglycemia and dehydration without significant ketoacidosis.
Test your knowledge with these 30 questions.
Question 1/30
Here are your results, .
Your Score
27/30
90%
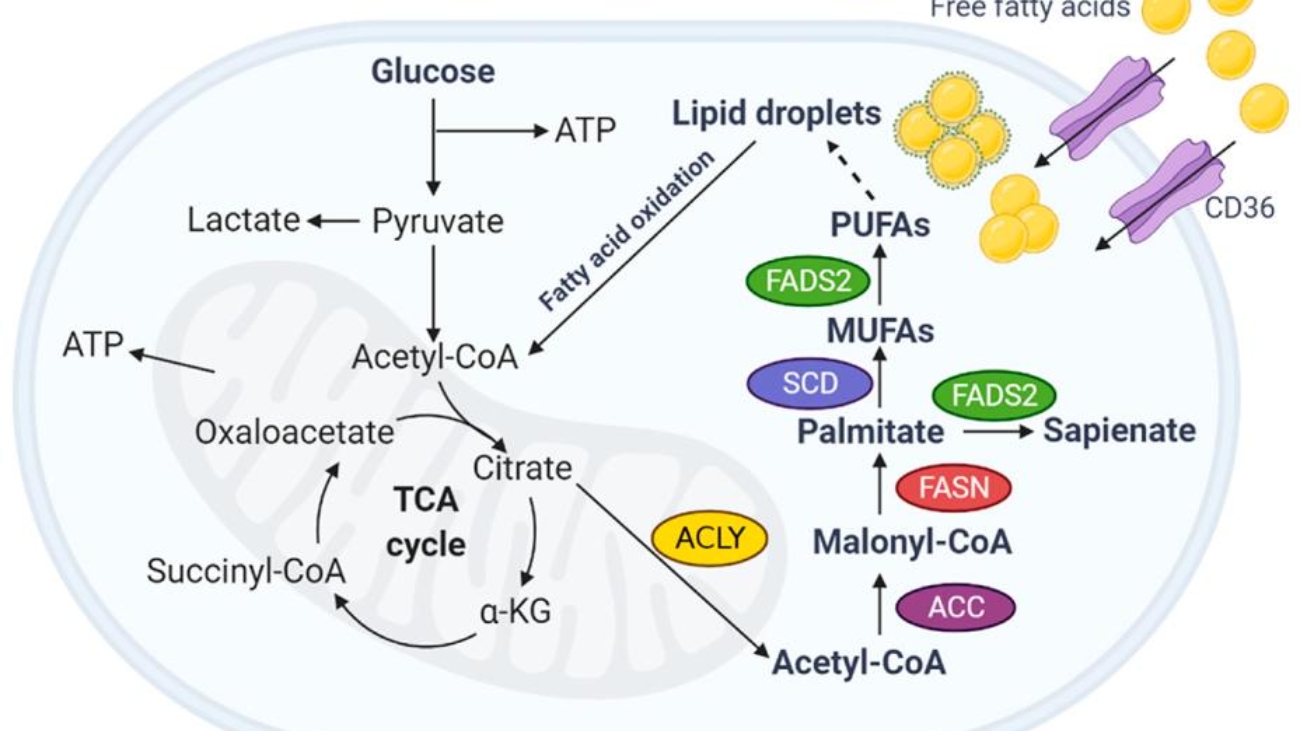
When energy is needed, stored triacylglycerols (TAGs) in adipose tissue must be broken down, and the resulting fatty acids transported to other tissues for oxidation.
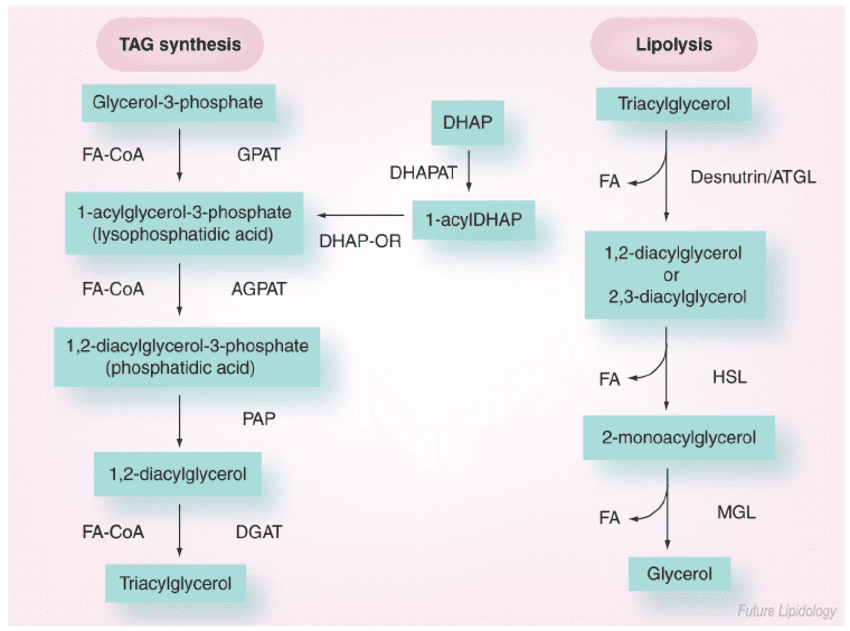
Lipolysis is the process of breaking down stored TAGs into fatty acids and glycerol, occurring in adipocytes.
Long-chain fatty acids are hydrophobic and require a carrier in the blood.
Long-chain fatty acids (LCFAs) cannot directly cross the inner mitochondrial membrane. They require the Carnitine Shuttle to enter the mitochondrial matrix for beta-oxidation.
Now, with the fatty acyl-CoA ready in the mitochondrial matrix, we can move on to the actual breakdown process: Fatty Acid Oxidation (Beta-Oxidation).
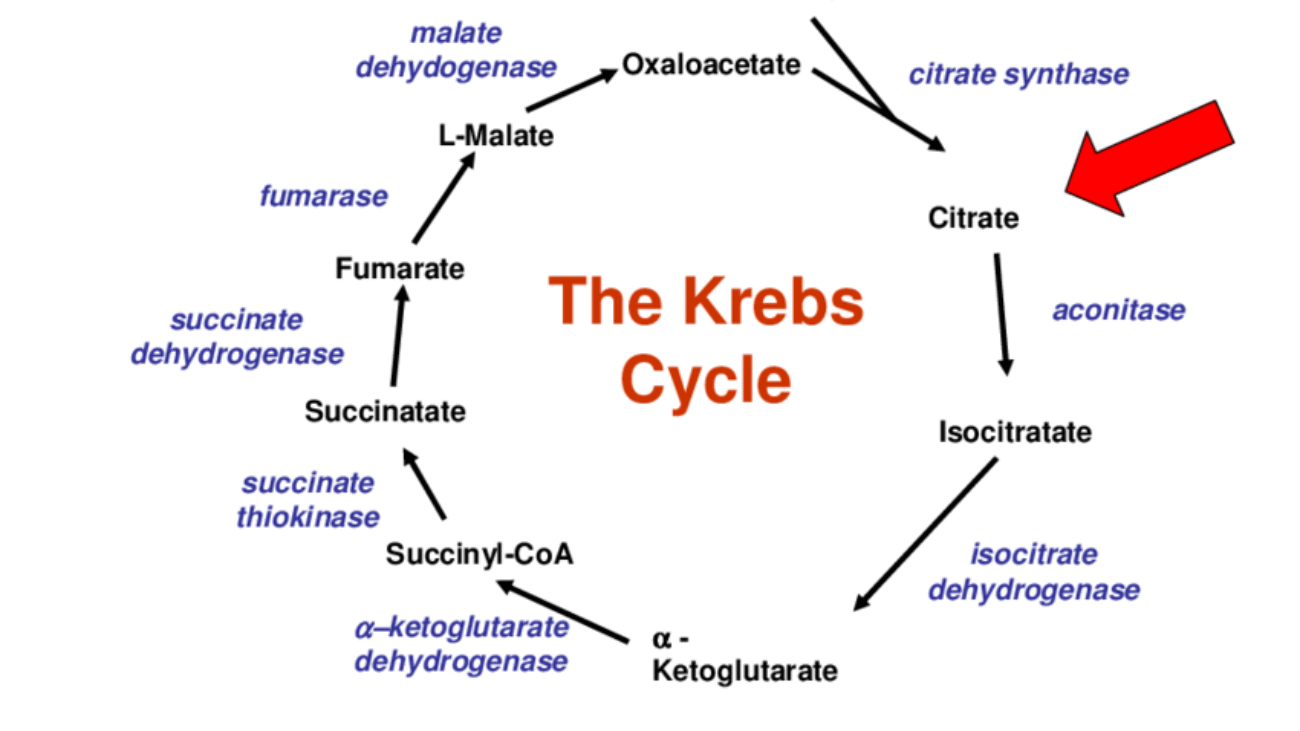
Once long-chain fatty acids (as fatty acyl-CoA) have successfully entered the mitochondrial matrix via the carnitine shuttle, they are ready for a cyclic process called β-oxidation. This pathway systematically cleaves two-carbon units from the carboxyl end of the fatty acyl-CoA, generating acetyl-CoA, NADH, and FADH₂, which then feed into the citric acid cycle and oxidative phosphorylation for ATP production.
Beta-oxidation is a four-step cyclic process. Each cycle shortens the fatty acyl-CoA by two carbons and produces one molecule of Acetyl-CoA, one NADH, and one FADH₂.
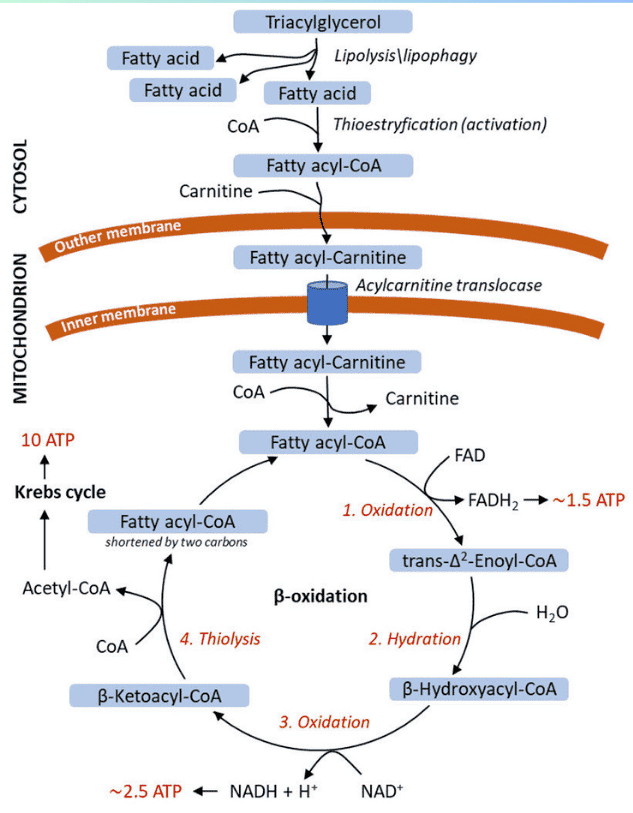
Input: Fatty Acyl-CoA (n carbons) → Output: 1 Acetyl-CoA + 1 FADH₂ + 1 NADH + Fatty Acyl-CoA (n-2 carbons)
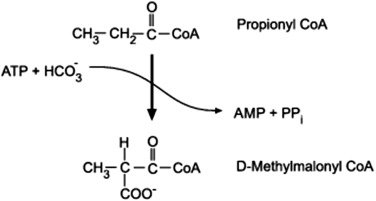
Under certain physiological conditions, particularly prolonged fasting, starvation, or uncontrolled diabetes, the liver produces significant amounts of ketone bodies from Acetyl-CoA. These ketone bodies serve as an alternative fuel source for extrahepatic (outside the liver) tissues, especially the brain, which cannot directly use fatty acids for energy.
Ketogenesis is stimulated when:
In essence, ketogenesis is a response to an oversupply of Acetyl-CoA (from fat breakdown) and an undersupply of OAA (due to gluconeogenesis) in the liver.
Ketogenesis occurs exclusively in the mitochondrial matrix of liver cells.
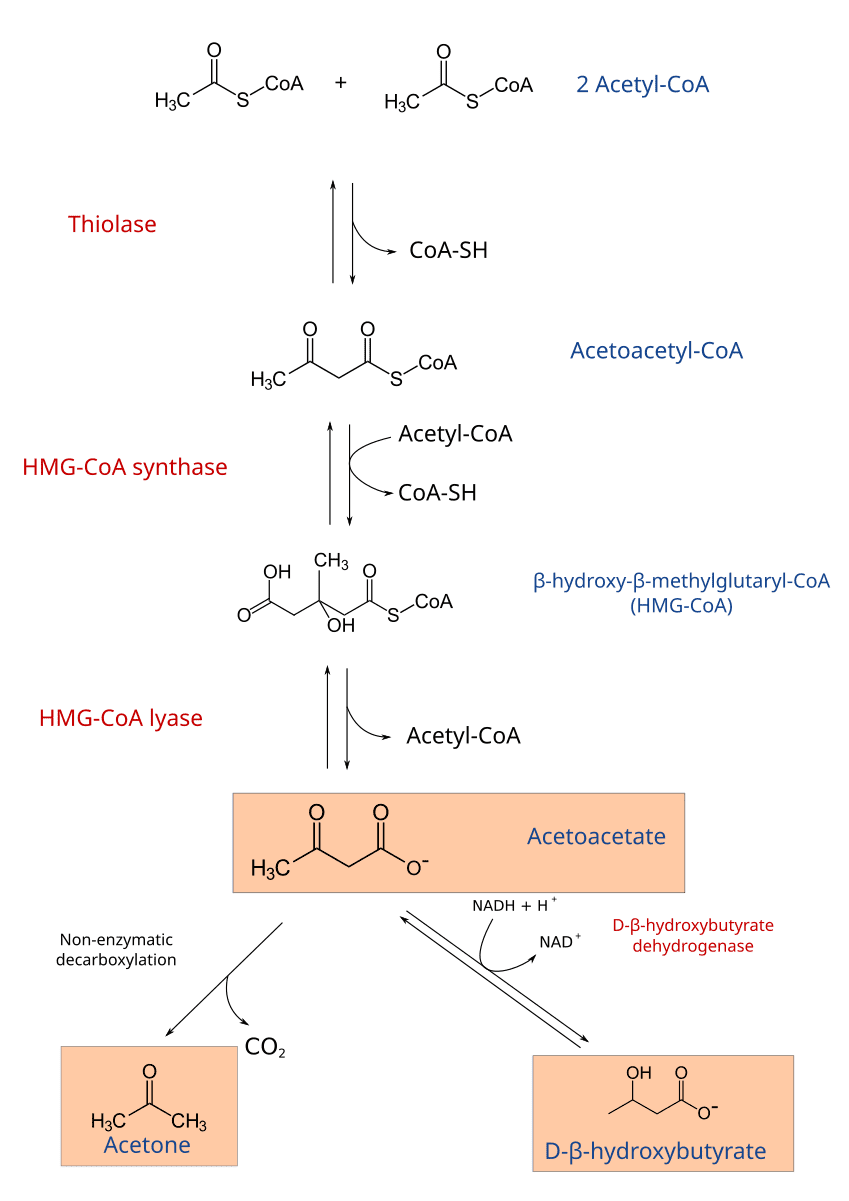
2 Acetyl-CoA → Acetoacetyl-CoA + CoA-SHAcetoacetyl-CoA + Acetyl-CoA + H₂O → β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) + CoA-SHHMG-CoA → Acetoacetate + Acetyl-CoAAcetoacetate + NADH + H⁺ ⇌ β-Hydroxybutyrate + NAD⁺Acetoacetate → Acetone + CO₂).Ketone bodies are water-soluble and can be transported via the bloodstream to peripheral tissues, which then convert them back into Acetyl-CoA for energy. The liver cannot utilize ketone bodies because it lacks a key enzyme for ketolysis.
Tissues that use Ketone Bodies: Brain, heart, skeletal muscle, renal cortex.
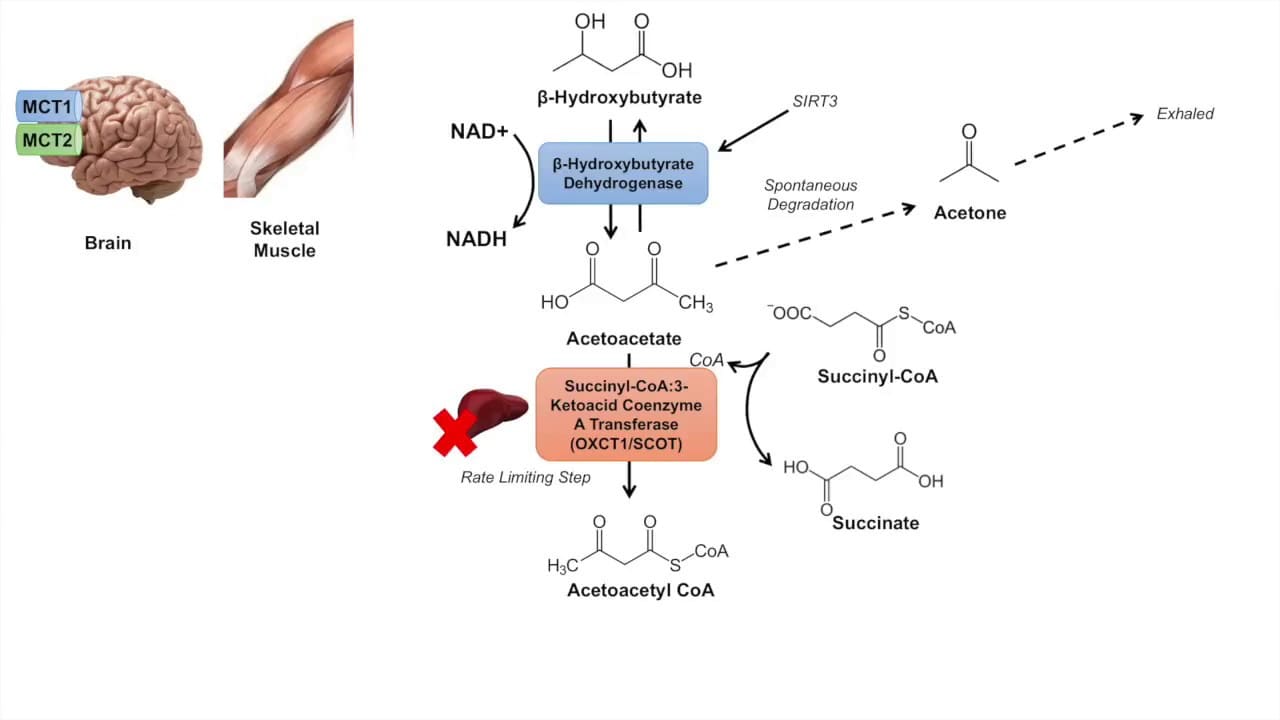
β-Hydroxybutyrate + NAD⁺ → Acetoacetate + NADH + H⁺Acetoacetate + Succinyl-CoA → Acetoacetyl-CoA + SuccinateAcetoacetyl-CoA + CoA-SH → 2 Acetyl-CoAThe 2 molecules of Acetyl-CoA produced can then enter the Citric Acid Cycle to generate ATP.
The production and utilization of ketone bodies are normally well-regulated. However, imbalances can lead to serious clinical conditions.
When the body has an abundance of energy, especially from a diet rich in carbohydrates, it converts excess glucose into fatty acids for long-term storage as triacylglycerols. This process is called lipogenesis.
Liver: The most active site of fatty acid synthesis.Adipose Tissue: Also synthesizes fatty acids.Lactating Mammary Glands: Synthesize fatty acids for milk production.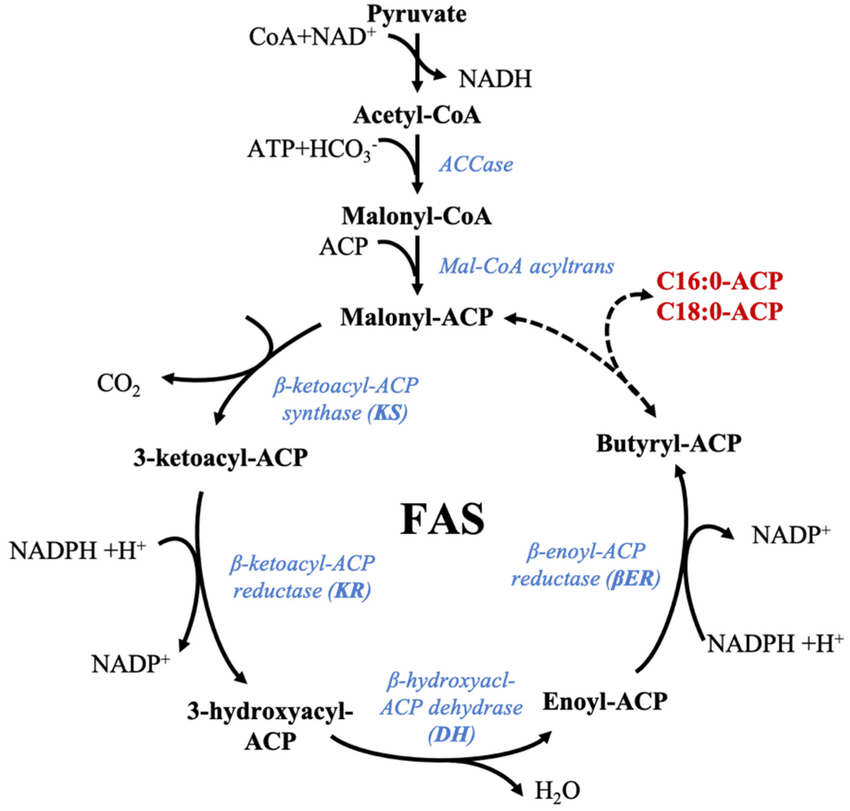
Fatty acid synthesis is essentially a reversal of β-oxidation, but it uses different enzymes, occurs in a different cellular compartment, and employs a different electron donor.
Citrate + ATP + CoA-SH → Acetyl-CoA + OAA + ADP + PiAcetyl-CoA + HCO₃⁻ + ATP → Malonyl-CoA + ADP + PiSynthesis is carried out by a multi-enzyme complex called Fatty Acid Synthase (FAS). It contains seven different enzymatic activities and an acyl carrier protein (ACP).
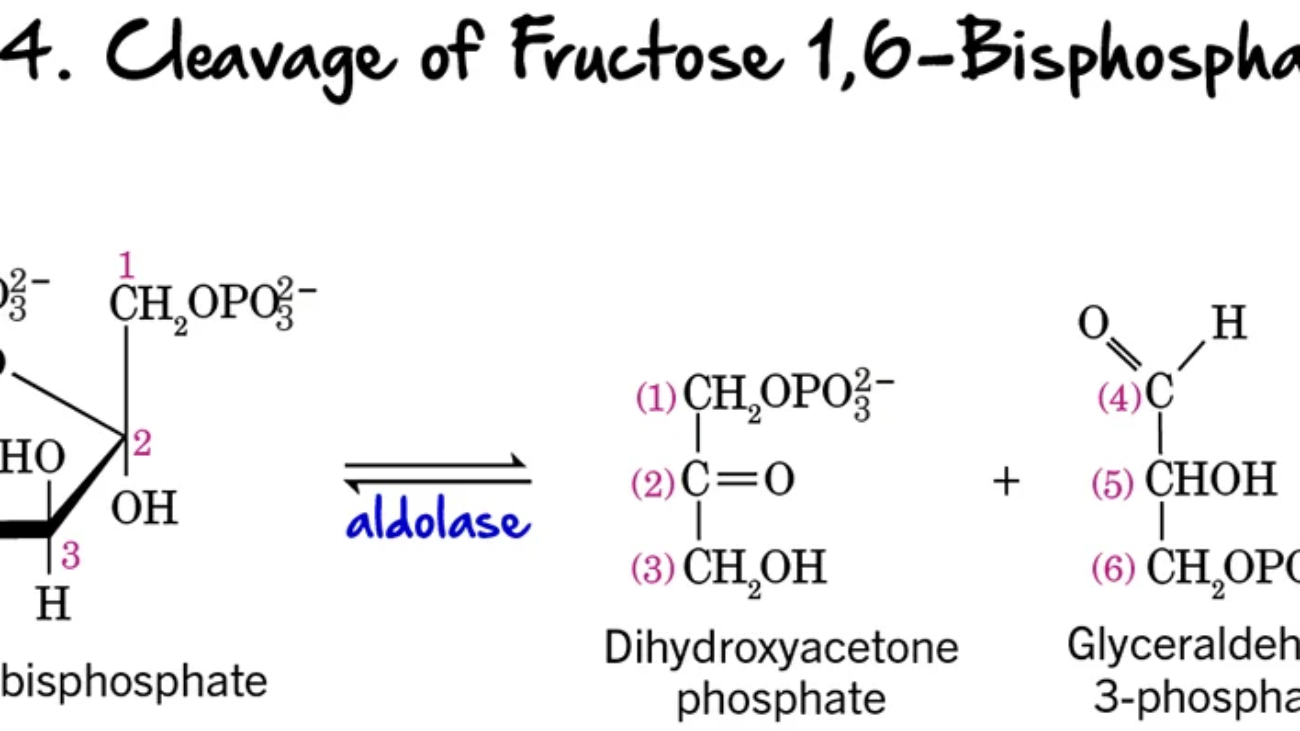
Each cycle adds a two-carbon unit from Malonyl-CoA and involves four steps:
After 7 cycles, the 16-carbon palmitoyl-ACP is formed and then released as free palmitate by a Thioesterase.
Overall Reaction: 8 Acetyl-CoA + 7 ATP + 14 NADPH → Palmitate + 8 CoA + 7 ADP + 7 Pi + 14 NADP⁺ + 6 H₂O
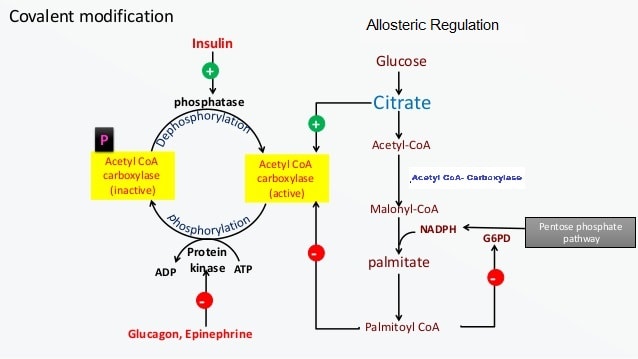
Once palmitate (16:0) is synthesized, it can be further modified:
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
The Pentose Phosphate Pathway (PPP), also known as the Hexose Monophosphate Shunt (HMP Shunt), is an alternative metabolic route for glucose metabolism that runs parallel to glycolysis. The HMP pathway is also known as the Warburg-Dickens pathway. About 10% of glucose entering this pathway per day. The liver & RBCs metabolise about 30% of glucose by this pathway.
Unlike glycolysis, its primary purpose is not to generate ATP. Instead, its main functions are:
Think of the PPP as a "shunt" because it diverts glucose-6-phosphate away from glycolysis to serve these distinct purposes, and can then feed intermediates back into glycolysis. It primarily occurs in the cytosol of cells.
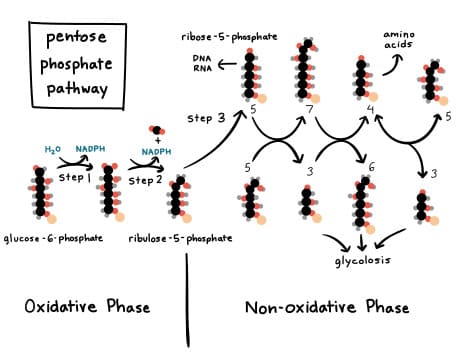
The Pentose Phosphate Pathway is divided into two distinct phases:
The PPP is critically important because it provides two essential molecules:
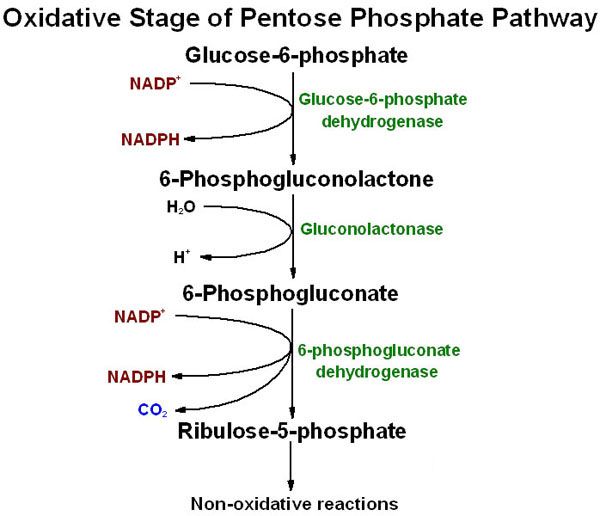
This phase consists of three main reactions, starting with glucose-6-phosphate and culminating in the production of NADPH and ribulose-5-phosphate.
The oxidative phase involves the following sequential reactions:
Glucose-6-phosphate + NADP⁺ → 6-Phosphogluconolactone + NADPH + H⁺6-Phosphogluconolactone + H₂O → 6-Phosphogluconate6-Phosphogluconate + NADP⁺ → Ribulose-5-phosphate + NADPH + H⁺ + CO₂The net reaction for the oxidative phase is:
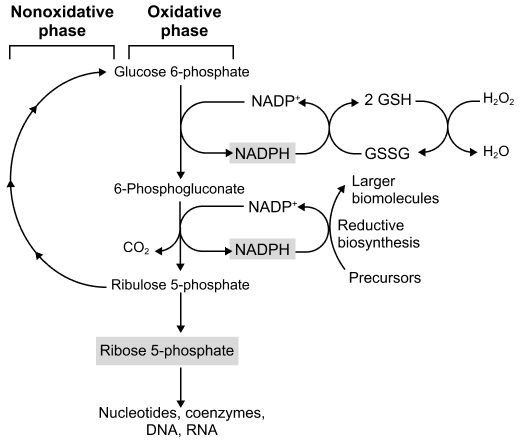
The non-oxidative phase is a series of reversible reactions that interconvert various sugar phosphates. Its primary functions are:
This phase involves three main enzymes: an isomerase, an epimerase, and two transketolases/transaldolases.
The ribulose-5-phosphate generated in the oxidative phase needs to be converted into other pentose sugars.
Ribulose-5-phosphate ⇌ Ribose-5-phosphateRibulose-5-phosphate ⇌ Xylulose-5-phosphateThese two enzymes are responsible for moving two-carbon and three-carbon units between sugar phosphates to produce glycolytic intermediates.
If 3 molecules of glucose-6-phosphate enter the oxidative phase, they produce 3 molecules of ribulose-5-phosphate and 6 NADPH. These 3 molecules of ribulose-5-phosphate are then processed through the non-oxidative phase:
These glycolytic intermediates can then enter glycolysis, be used for gluconeogenesis, or be recycled to continue the PPP.
The reversibility of the non-oxidative phase is key, allowing the pathway to operate in different modes:
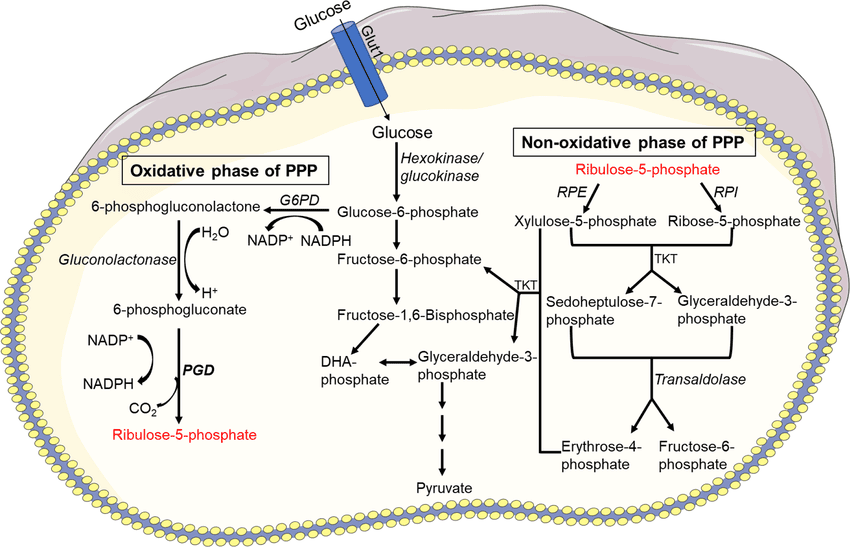
The activity of the PPP varies significantly among different tissues, directly reflecting their metabolic demands for its key products: NADPH and ribose-5-phosphate.
The liver is a central metabolic hub with a high demand for NADPH for:
Adipocytes are specialized for fat storage and have a very high demand for NADPH to support the massive amount of fatty acid synthesis that occurs here.
RBCs lack mitochondria and are constantly exposed to oxidative stress. The PPP is their only source of NADPH for antioxidant defense, used to maintain reduced glutathione (GSH) and protect the cell.
Tissues like the adrenal cortex, testes, and ovaries are primary sites of steroid hormone synthesis and have a high demand for NADPH for these hydroxylation reactions.
During lactation, the mammary gland synthesizes large amounts of fatty acids for milk production, requiring a high supply of NADPH.
Tissues like bone marrow, skin, intestinal mucosa, and tumors are continuously proliferating and require constant DNA and RNA synthesis. They have a high demand for ribose-5-phosphate for nucleotide synthesis.
The non-oxidative phase can be reversed in these cells to primarily produce ribose-5-phosphate from glycolytic intermediates.
Test your knowledge with these 35 questions.
Question 1/35
Here are your results, .
Your Score
33/35
94%