Biochemistry: Enzymes Exam
Test your knowledge with these 30 questions.
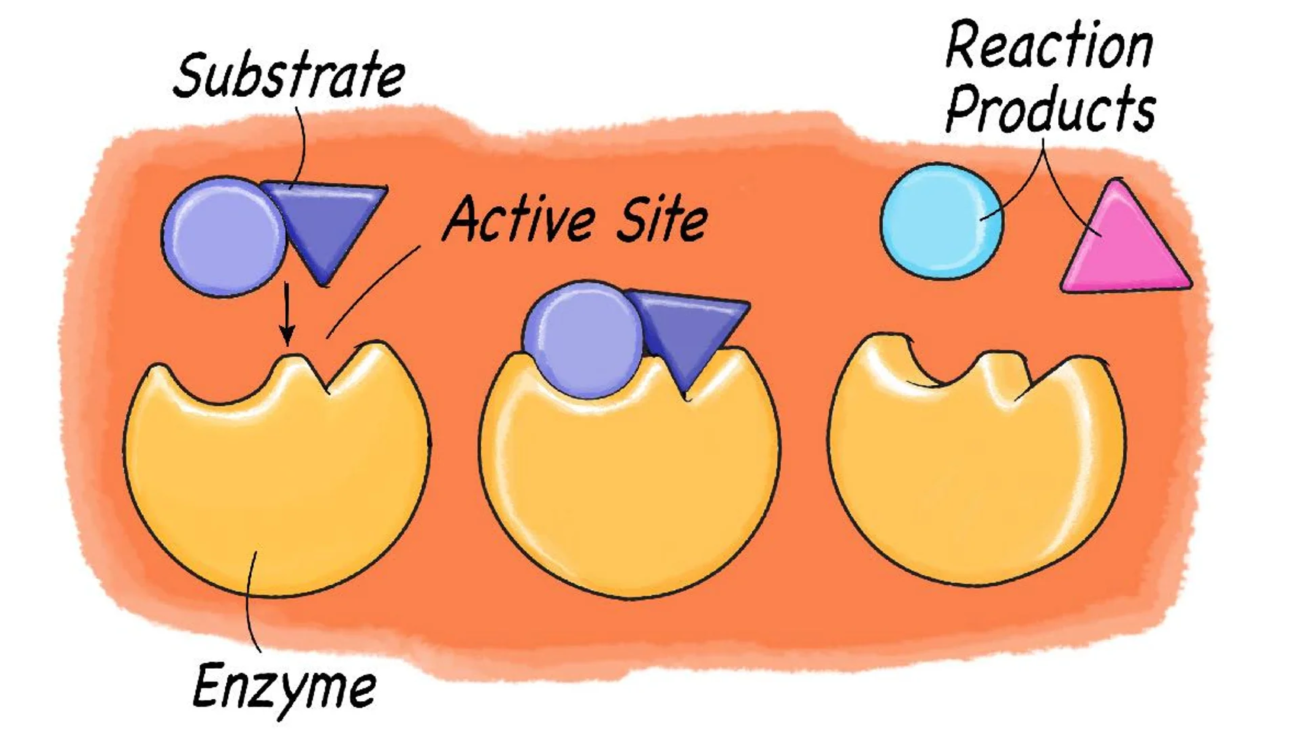
Enzymes Exam
Question 1/30
Exam Complete!
Here are your results, .
Your Score
28/30
93%
Test your knowledge with these 30 questions.
Question 1/30
Here are your results, .
Your Score
28/30
93%
A peptide or protein sequence is the specific linear order of amino acids linked together by peptide bonds. There are very specific conventions for how these sequences are written and read, which are essential for clear, unambiguous communication in biochemistry and molecular biology.
Every peptide or polypeptide chain exhibits a distinct directionality, meaning it has a defined "start" and an "end." This intrinsic polarity is fundamental to how proteins are synthesized, fold, and function.
Peptide sequences are always read from left to right, starting from the N-terminus and proceeding sequentially towards the C-terminus.
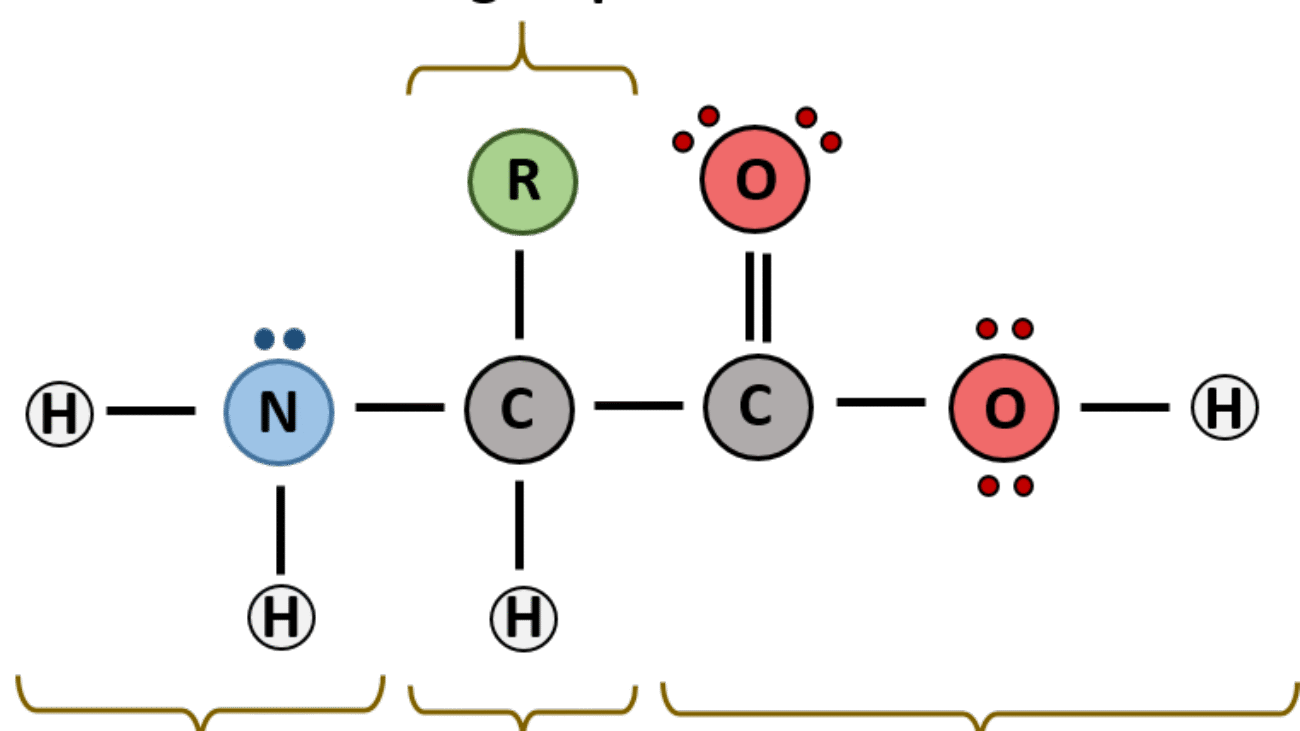
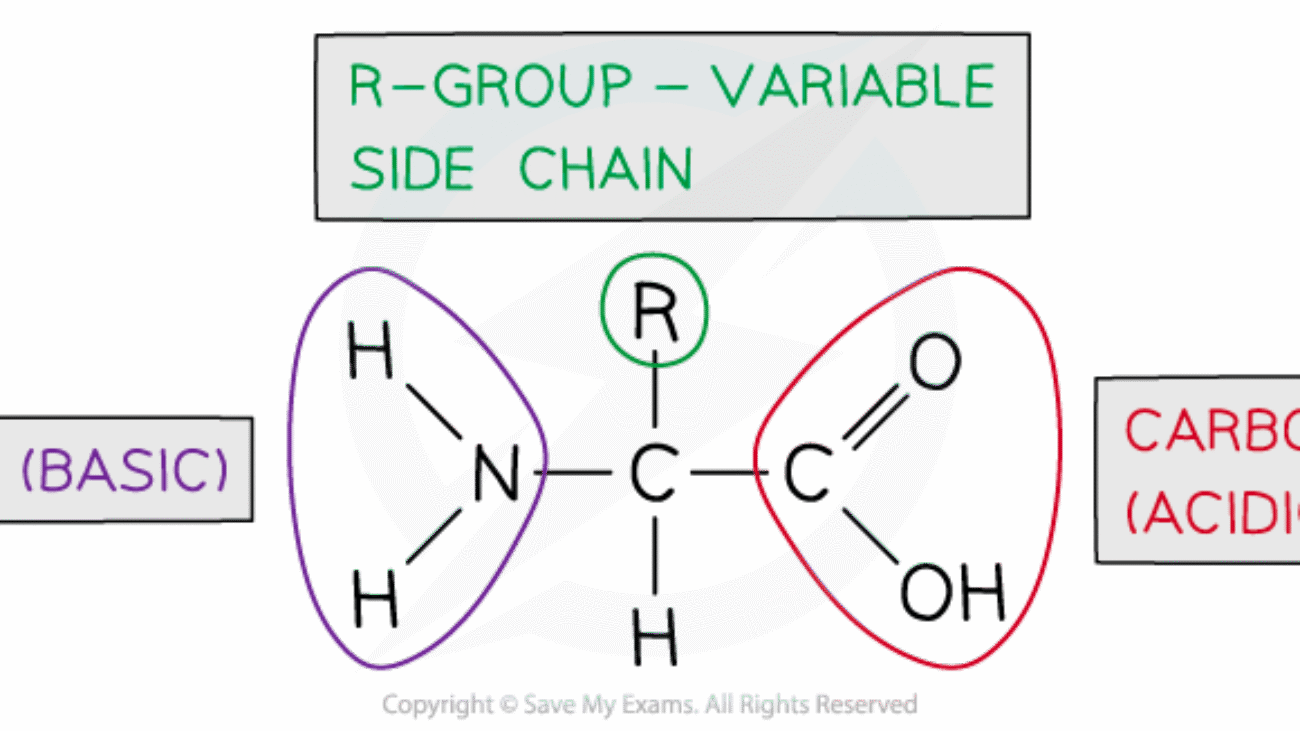
Each amino acid unit within the peptide chain, after forming peptide bonds, is referred to as an amino acid residue. This term emphasizes that each amino acid has lost the elements of water (a hydrogen atom from its amino group and a hydroxyl group from its carboxyl group) when participating in the formation of a peptide bond. Within the chain, only the R-group and the α-carbon, along with parts of the backbone, remain.
To simplify the writing and reading of often very long protein sequences, standard abbreviations are universally used for the 20 common genetically encoded amino acids:
| Amino Acid | Three-Letter Code | One-Letter Code |
|---|---|---|
| Alanine | Ala | A |
| Arginine | Arg | R |
| Asparagine | Asn | N |
| Aspartate | Asp | D |
| Cysteine | Cys | C |
| Glutamine | Gln | Q |
| Glutamate | Glu | E |
| Glycine | Gly | G |
| Histidine | His | H |
| Isoleucine | Ile | I |
| Leucine | Leu | L |
| Lysine | Lys | K |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Proline | Pro | P |
| Serine | Ser | S |
| Threonine | Thr | T |
| Tryptophan | Trp | W |
| Tyrosine | Tyr | Y |
| Valine | Val | V |
Note: For cases where the exact amide status is unknown or ambiguous:
When asked to "name" a peptide or write its sequence, you list the amino acid residues in order from the N-terminus to the C-terminus, using their standard abbreviations.
Let's carefully examine this peptide structure to determine its sequence.
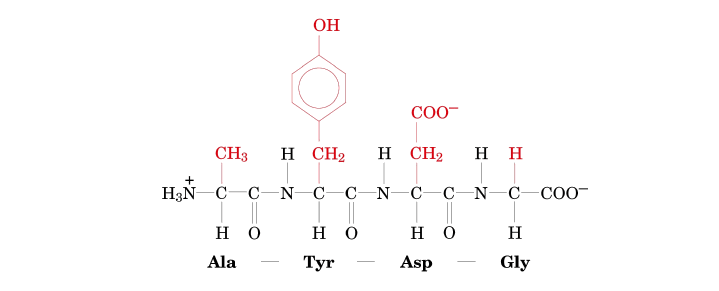
Step-by-step identification:
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%

Test your knowledge with these 51 questions.
Question 1/51
Here are your results, .
Your Score
48/51
94%
Test your knowledge with these 30 questions.
Question 1/30
Here are your results, .
Your Score
28/30
93%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
Test your knowledge with these 40 questions.
Question 1/40
Here are your results, .
Your Score
38/40
95%
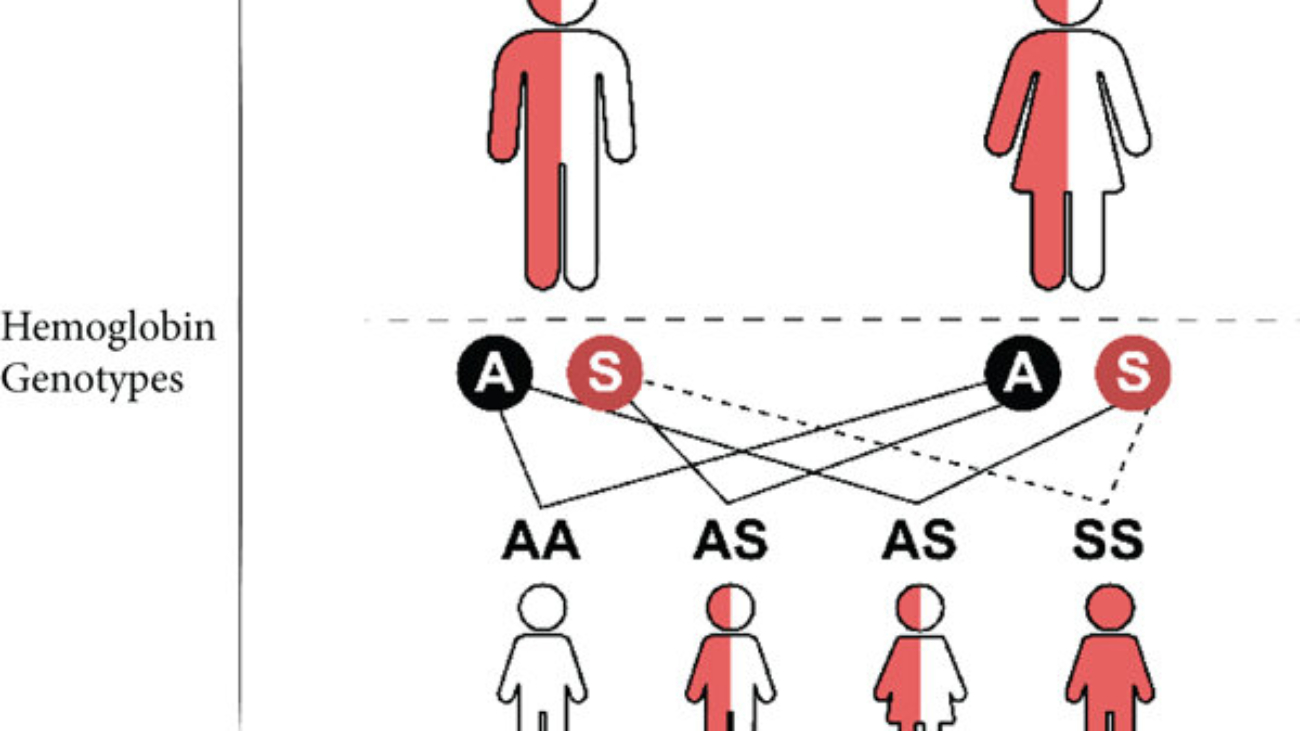
A 2-year-old boy from Mukono district presents with recurrent episodes of severe bone pain (hands, feet, and sternum pain), jaundice, and fatigue for 3 days.
Laboratory findings reveal:
A diagnosis of Vaso-occlusive crisis, and severe anaemia in Sickle Cell Disease was made.
This part requires a detailed breakdown of the specific molecular error in the patient's haemoglobin protein, focusing on the identity of the amino acids and the genetic origin of the mistake.
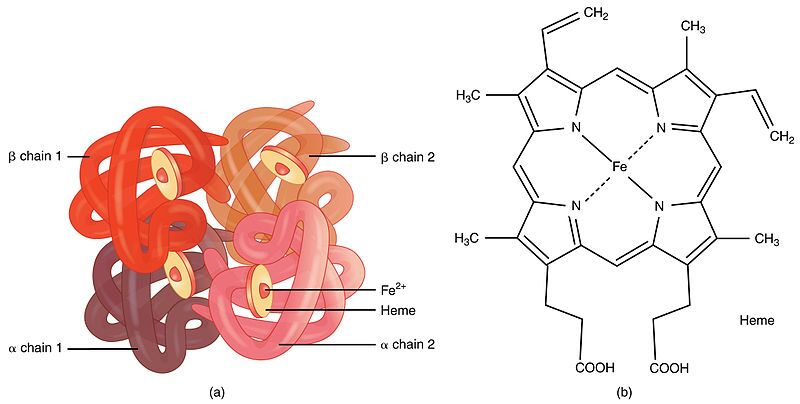
First, it's important to understand what haemoglobin is. Haemoglobin is the primary protein found within red blood cells (erythrocytes) and its main function is to transport oxygen from the lungs to the body's tissues. It is a large, complex protein with a quaternary structure, meaning it is composed of multiple polypeptide subunits. A normal adult haemoglobin molecule (HbA) is a tetramer, consisting of four chains: two identical alpha (α)-globin chains and two identical beta (β)-globin chains. The genetic defect in sickle cell disease specifically affects the gene that provides the instructions for the beta-globin chain.

The defining molecular event in sickle cell disease is a single amino acid substitution at a precise location within the beta-globin polypeptide chain.
In a person with normal adult haemoglobin (HbA), the amino acid at the sixth position from the beginning (the N-terminus) of the beta-globin chain is Glutamic Acid (abbreviated as Glu or E).
In this patient with sickle cell disease, the haemoglobin is abnormal (called HbS). At that exact same sixth position, the Glutamic Acid has been replaced by the amino acid Valine (abbreviated as Val or V).
This single change, Glu6Val, is the sole cause of the disease.

The severity of this substitution is due to the drastically different chemical "personalities" of the R-groups (side chains) of Glutamic Acid and Valine. This position is on the outer surface of the protein, where it is exposed to the watery environment inside the red blood cell.
| Amino Acid | Chemical Class & Properties | Behavior in Water |
|---|---|---|
| Glutamic Acid (Normal) | Its side chain contains a carboxyl group (`-CH₂-CH₂-COOH`). At the neutral pH inside a red blood cell (~7.4), this group loses a proton and becomes negatively charged (`-COO⁻`). Therefore, it is an acidic, polar, and charged amino acid. | Because it is charged and polar, Glutamic Acid is hydrophilic ("water-loving"). It forms favorable interactions with polar water molecules and is perfectly stable on the protein's surface. |
| Valine (Mutant) | Its side chain is an isopropyl group (`-CH(CH₃)₂`), which is a small, branched structure made only of carbon and hydrogen. These bonds are nonpolar. Therefore, Valine is a nonpolar, aliphatic, and neutral amino acid. | Because it is nonpolar, Valine is hydrophobic ("water-fearing"). It is thermodynamically unfavorable for this "oily" side chain to be exposed to water. It will seek to interact with other nonpolar groups to hide from the aqueous environment. |
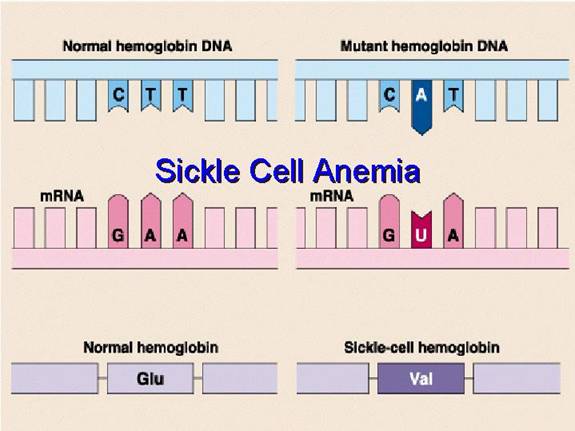
This amino acid error originates from a single change in the DNA sequence of the beta-globin gene. This type of mutation is called a point mutation, specifically a missense mutation because it results in a codon that codes for a different amino acid.
Therefore, a single DNA base change leads to a single mRNA codon change, which in turn leads to the single, catastrophic amino acid substitution that defines sickle cell disease.
This section explains the step-by-step process of how the single Glu6Val substitution causes the haemoglobin to malfunction and leads to the patient's observed symptoms.

The key event is the behavior of HbS when it is in the deoxygenated state. In the oxygenated state (in the lungs), HbS functions almost normally as an oxygen carrier.
Shape Distortion: These long, stiff haemoglobin polymers grow to be longer than the diameter of the red blood cell itself. They physically push against the cell membrane from the inside, distorting the cell from its normal, flexible biconcave disc shape into a rigid, elongated, crescent or "sickle" shape.
Loss of Deformability: This sickling process causes a dramatic loss of the cell's flexibility. It becomes hard and unable to deform. This process is initially reversible if the cell becomes reoxygenated, but repeated sickling events cause permanent membrane damage, leading to irreversibly sickled cells.
The physical properties of these sickled cells are directly responsible for the patient's symptoms:
Knowing that the core problem is a hydrophobic amino acid causing polymerization allows for the design of targeted therapies.
This approach aims to reduce the relative concentration of the problematic HbS.
This is the most direct chemical approach, aiming to stop the Valine from interacting with its target.
This is the most fundamental approach, aiming to fix the DNA instruction so the correct amino acid is made.

Test your knowledge with these 25 questions.
Question 1/25
Here are your results, .
Your Score
23/25
92%
Test your knowledge with these 51 questions.
Question 1/51
Here are your results, .
Your Score
48/51
94%

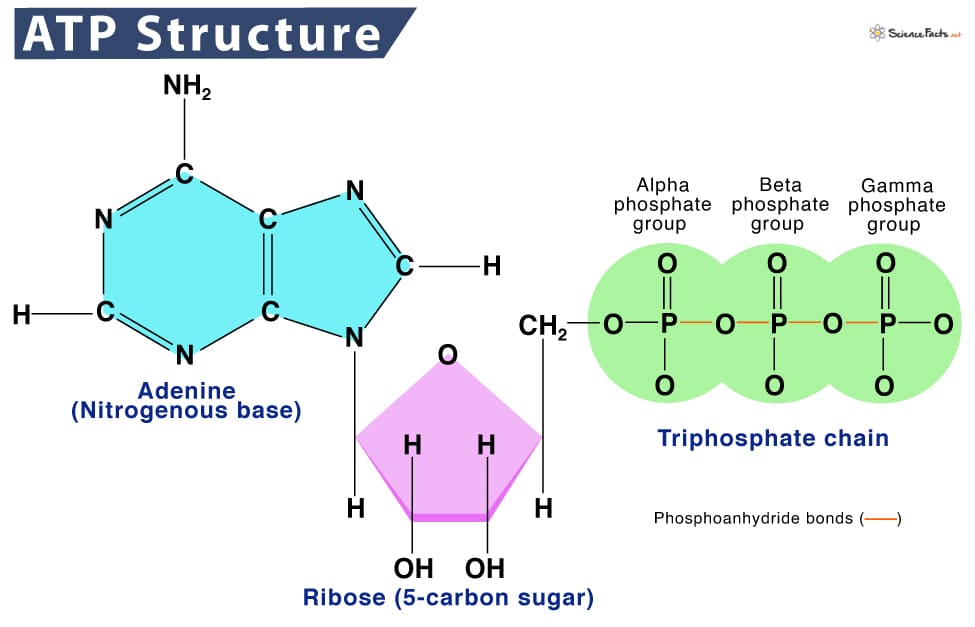
Once the body gets energy from food, it doesn't directly use these complex food molecules to power every single tiny process. Instead, the body converts the chemical energy stored in these food molecules into a much more manageable and readily available form: a special molecule called ATP.
Why is ATP called the "Energy Currency"? Think of it like money. You don't get paid in raw materials; you get paid in money, which you can use to buy whatever you need. Similarly, your body converts energy from diverse food sources into ATP (the "money"). Then, it uses ATP to "pay for" all its energy-requiring processes.
ATP is the direct, usable form of energy for almost all cellular activities.
The key lies in the "high-energy" chemical bonds connecting its three phosphate groups. When your cells need energy, they break off one of the phosphate groups from ATP. This breaking of the bond releases a significant amount of free energy that the cell can immediately use.
ATP → ADP + Pᵢ + Energy
This reaction is reversible. When your body has excess energy, it can use it to reattach the phosphate group to ADP, converting it back into ATP, thus "recharging the battery."
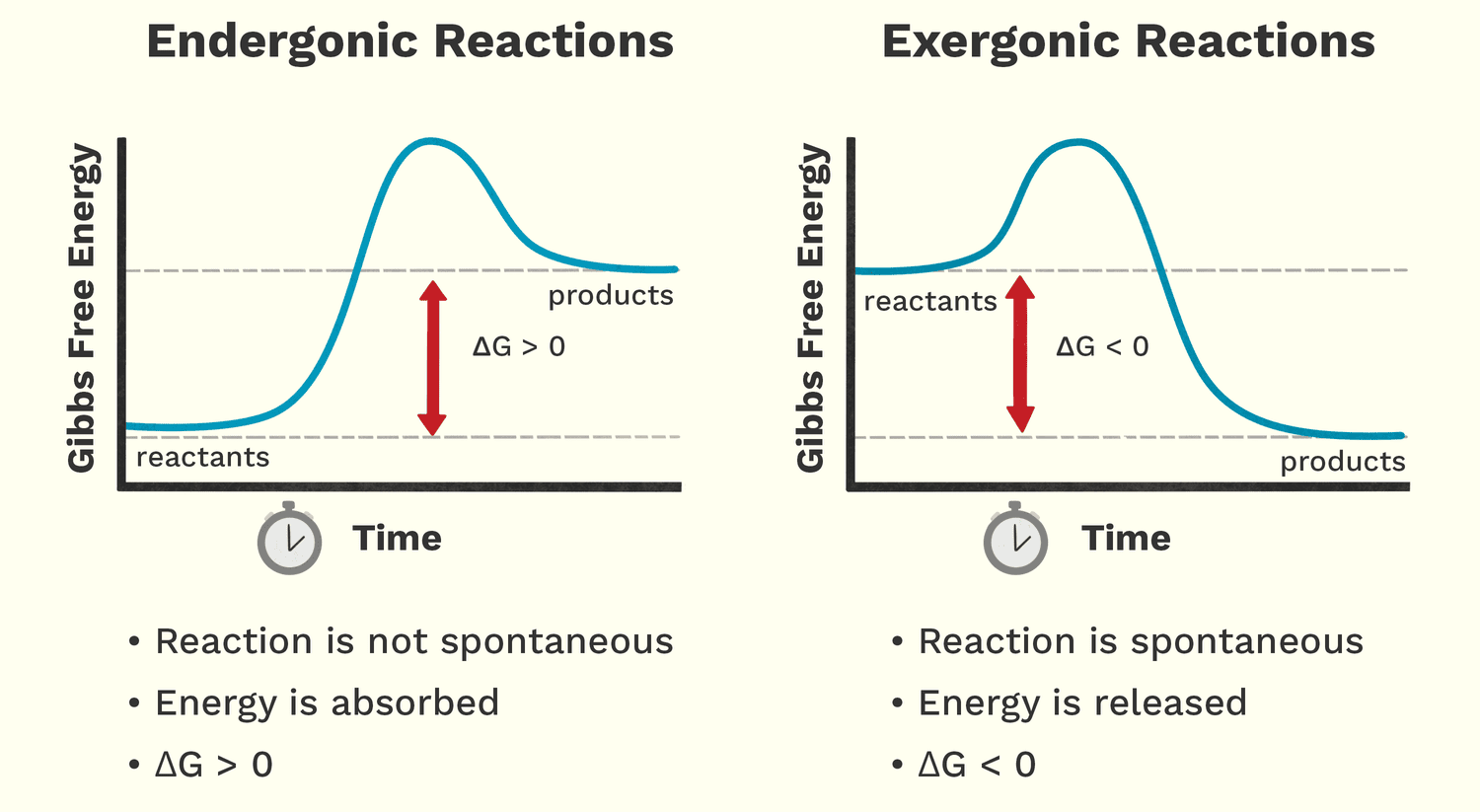
Free energy (Gibbs Free Energy, G) is the amount of energy available to do work within a system. It helps us predict whether a chemical reaction will happen spontaneously (release energy) or require an input of energy.
Analogy: A person moving down a hill is a spontaneous process that releases energy. A person lifting a weight up a hill is a non-spontaneous process that requires energy.
These reactions release free energy and can happen spontaneously. The change in free energy (ΔG) is negative (ΔG < 0).
Biological Examples:
These reactions require an input of free energy and are non-spontaneous. The change in free energy (ΔG) is positive (ΔG > 0).
Biological Examples:
Life thrives by ingeniously linking these two types of reactions together. Cells use the energy released from an exergonic reaction (like ATP breaking down) to drive an endergonic reaction that needs energy. This is called energy coupling. ATP is the perfect intermediate, acting as the bridge that carries energy from energy-releasing pathways to energy-requiring processes.

The overarching scientific field that governs all energy concepts is Thermodynamics. It is a branch of science that deals with the transformation or interconversion of different forms of energy, and how that energy is utilized.
Literally, thermodynamics is about the power of heat or the movement of heat and energy. While "heat" is in the name, it encompasses all forms of energy relevant to biological systems, including light, thermal, chemical, electrical, and mechanical energy.
Thermodynamics is built upon a few fundamental principles known as the Laws of Thermodynamics. These laws are absolute and govern all energy transformations in the universe, including those happening inside the human body.
"Two systems in equilibrium with a third system are in thermal equilibrium with each other."
Meaning: This law defines temperature and is the principle that allows a thermometer to accurately measure a patient's temperature.
Biological Implication: This law underpins the concept of body temperature and thermoregulation. Our bodies constantly strive to maintain a thermal equilibrium (homeostasis).
"Energy cannot be created or destroyed, only transformed from one form to another."
Meaning: The total amount of energy in the universe is constant. You can't get something for nothing.
Biological Implications: Plants don't "make" energy; they transform light energy into chemical energy. When you exercise, you convert chemical energy from food into mechanical energy and heat. Life needs a constant input of energy because organisms are continuously transforming it from external sources to fuel internal processes.
"In any isolated system, the total entropy (disorder) can only increase or remain constant."
Meaning: The universe naturally tends towards a state of greater disorder, randomness, or chaos. Things naturally fall apart; they do not spontaneously become more organized without external effort.
Biological Implications: Living organisms are incredibly complex, highly ordered structures. To maintain this order and fight against entropy, organisms must constantly consume energy. Life is a continuous battle against the Second Law. Every energy transformation results in some energy being "lost" as unusable heat, increasing the entropy of the environment.
"The entropy of a system approaches a constant minimum value as its temperature approaches absolute zero."
Meaning: As a system's temperature gets closer to absolute zero (-273.15 °C), the disorder of the system approaches a minimum. At absolute zero, a perfect crystal would theoretically have zero entropy (perfect order).
Biological Implication: This law highlights the relationship between temperature and molecular motion/disorder. Very low temperatures reduce molecular motion, which is why cryopreservation attempts to halt metabolic processes by drastically reducing temperature and entropy.
Bioenergetics is essentially the application of thermodynamic principles to biological systems. It helps us understand:
These laws have direct clinical applications:
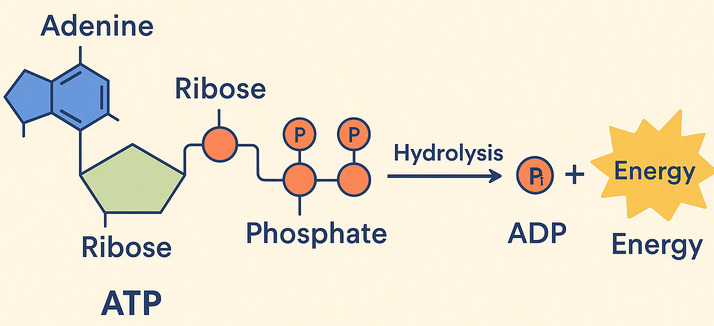
We've already introduced ATP as the energy currency that cells use to "pay for" their work. Now let's understand exactly how this remarkable molecule functions in this essential role.
ATP (Adenosine Triphosphate) is made of adenosine and three phosphate groups. The key to its power lies in the bonds between these phosphate groups, often called "high-energy phosphate bonds."
The term "high-energy" refers to the fact that when these bonds are broken, a significant amount of free energy is readily released. This is because the three negatively charged phosphate groups strongly repel each other, creating strain. Breaking the bond reduces this repulsion, and the remaining molecules (ADP and Pᵢ) settle into a more stable, lower-energy state. The difference in energy is what the cell can harness.
When the cell needs energy, it breaks the outermost phosphate bond in a process called hydrolysis, because a molecule of water (H₂O) is used to break the bond.
ATP + H₂O → ADP + Pᵢ + Free Energy
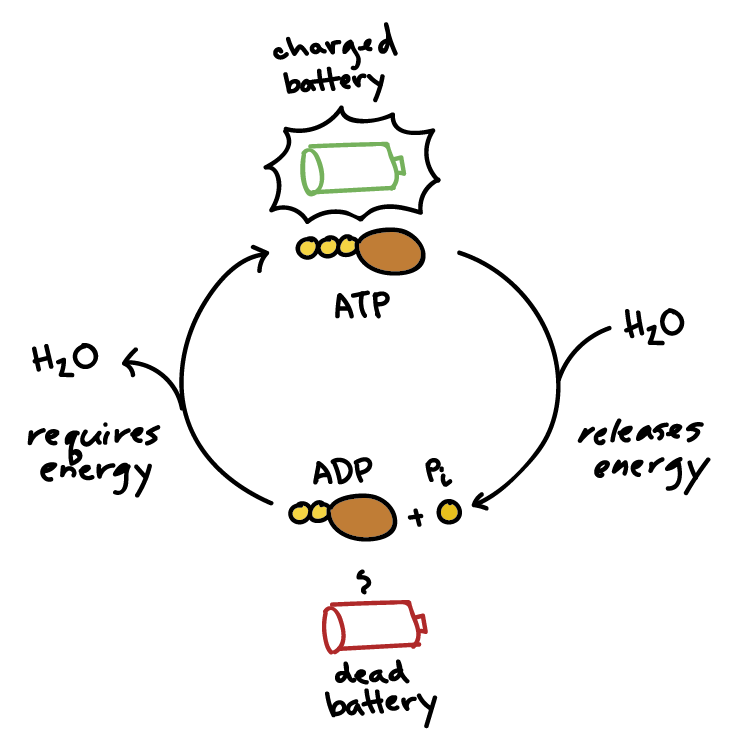
This is truly the magic of ATP! It perfectly acts as the bridge between energy-releasing (exergonic) and energy-requiring (endergonic) processes.
Life depends on a continuous, rapid cycle of ATP breakdown and synthesis:
ADP + Pᵢ + Energy (from food) → ATP + H₂OATP + H₂O → ADP + Pᵢ + Free Energy (for work)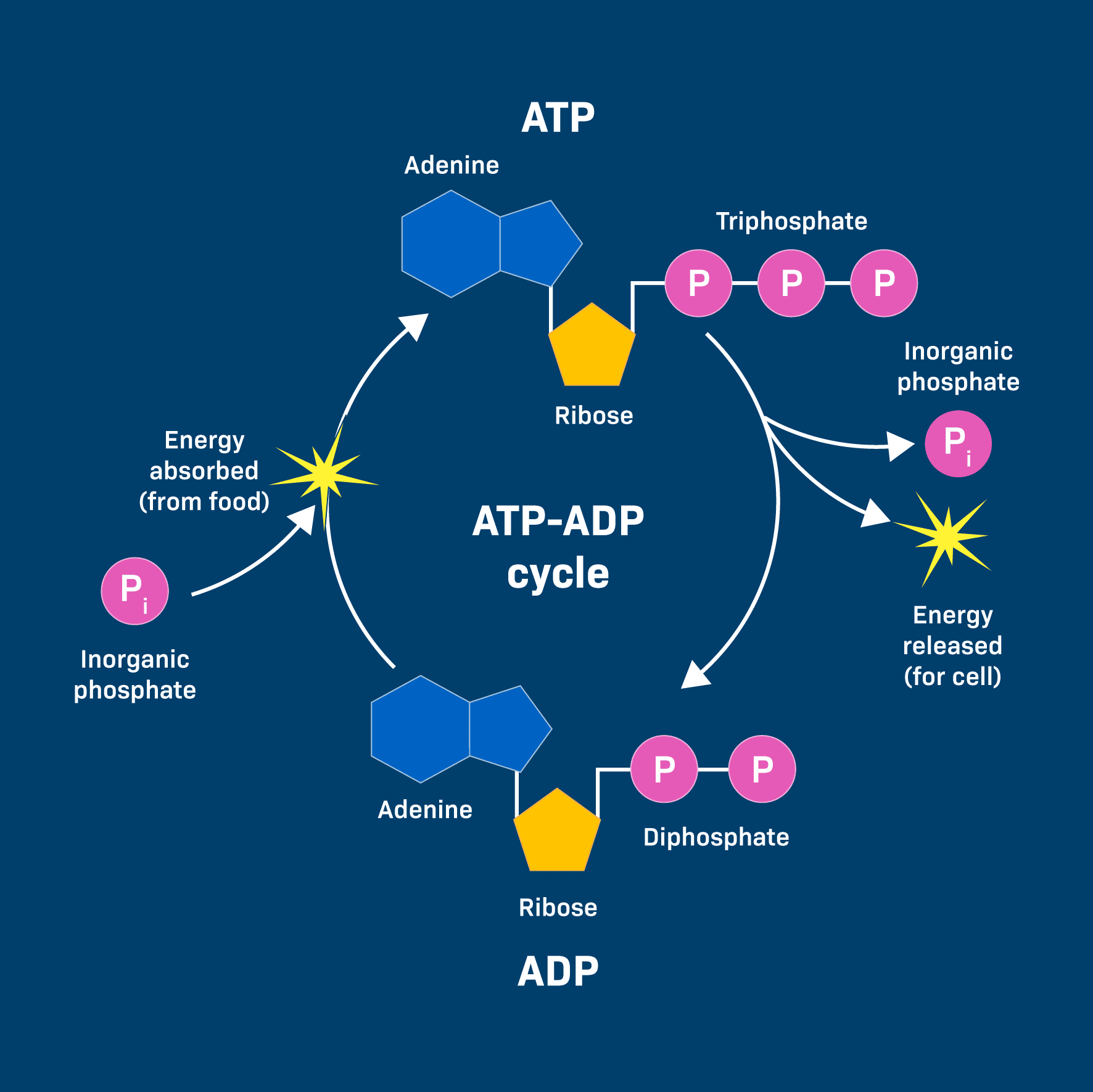
This energy is often transferred through a clever mechanism called phosphorylation. The phosphate group released from ATP is temporarily transferred to another molecule. This energizes the receiving molecule, making it more reactive and priming it to undergo its desired endergonic reaction.
Example: Muscle Contraction
An ATP molecule binds to a muscle protein (myosin). The ATP is hydrolyzed, and the phosphate (Pᵢ) temporarily attaches to the protein (phosphorylation). This causes a change in the protein's shape, leading to the physical contraction (the "work").
So, to summarize the continuous flow of energy that powers life:
Understanding ATP's role is fundamental to comprehending cellular health:

We previously touched upon the Second Law of Thermodynamics, which introduced the powerful idea that things naturally tend towards disorder. This concept is called entropy, and it's a critical component of understanding where "free energy" comes from.
Entropy (S) is a fundamental thermodynamic property that serves as a quantitative measure of randomness or disorder within a system. The more ways particles can be arranged, or the more freely they can move, the higher the entropy.
Analogy: Generally, gases (high entropy, chaotic) have higher entropy than liquids (medium entropy, less ordered), which have higher entropy than solids (low entropy, ordered). Breaking large, complex molecules into smaller, simpler ones also increases entropy.
Enthalpy (H) is essentially the total heat content or the total potential energy contained within a system at constant pressure. We are most interested in the change in enthalpy (ΔH).
This powerful equation is the heart of bioenergetics because it connects these concepts to determine whether a reaction will be spontaneous (exergonic) or require energy (endergonic).
Reactions are most likely to be spontaneous (exergonic) if they release heat (negative ΔH) AND increase disorder (positive ΔS).
6CO₂ + 6H₂O + Light → C₆H₁₂O₆ + 6O₂
C₆H₁₂O₆ + 6O₂ → 6CO₂ + 6H₂O + Energy
We've talked about ATP hydrolysis as releasing energy, but how does that energy actually get used? The primary way is through phosphoryl group transfer, often referred to simply as phosphorylation.
A phosphoryl group transfer is the movement of a phosphate group (Pᵢ) from one molecule to another. ATP is the most common donor. The enzyme-catalyzed transfer of the terminal phosphate group from ATP to a recipient molecule results in a phosphorylated recipient and ADP.
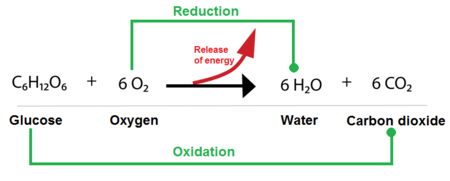
While phosphoryl group transfers are about using energy, oxidation-reduction (redox) reactions are primarily about harvesting and transferring energy from nutrient molecules. This is how cells extract energy from food.
These are always coupled reactions:
A helpful mnemonic is LEO the lion says GER! (Lose Electrons Oxidation, Gain Electrons Reduction).
In biological systems, the transfer of electrons often happens along with the transfer of protons (H⁺), so oxidation often means losing hydrogen atoms (dehydrogenation), and reduction often means gaining them (hydrogenation).
Cells use specialized molecules to pick up and carry electrons. The two most important are:
Test your knowledge with these 20 questions.
Question 1/20
Here are your results, .
Your Score
18/20
90%