Drug Interactions & Loss of Effect
By the time you finish studying this guide, you will be absolute masters of the following concepts:
- Describing Drug Combinations: You will know exactly what happens when doctors prescribe two drugs at the same time. You will understand the mathematical and biological differences between additive, synergistic, potentiation, and antagonistic interactions.
- Explaining the Loss of Drug Effect: You will understand the exact biological reasons why a drug might stop working in a patient over time. You will be able to clearly differentiate between tachyphylaxis, tolerance, refractoriness, and drug resistance.
Part 1: Combining Drugs with Similar or Related Effects
In clinical medicine, patients rarely take just one medication. When two or more drugs are given concurrently (at the same time), they interact. These interactions can be highly beneficial (helping the patient heal faster) or highly dangerous. We classify these outcomes into four specific mathematical categories based on how their dose-response curves interact.
Math Rule: 1 + 1 = 2
An additive effect occurs when the combined effect of two drugs equals the exact sum of their individual effects. Neither drug boosts the other; they just work side-by-side doing their own job, often acting on similar receptors or pathways.
- Clinical Example: Aspirin + Paracetamol (Acetaminophen). Both of these medications relieve pain and reduce fever. If you take 50% of a full dose of Aspirin and 50% of a full dose of Paracetamol together, you get exactly 100% pain relief.
- Expanded Mechanism: Aspirin works predominantly in the peripheral tissues by irreversibly inhibiting COX-1 and COX-2 enzymes. Paracetamol works more centrally in the brain (possibly via COX-3 or peroxidase sites). Their actions simply add up.
- Clinical Knowledge: Why do doctors do this? By combining two drugs, the doctor can use a lower dose of each individual drug. This drastically reduces the risk of dose-dependent side effects (like severe stomach bleeding from too much Aspirin, or fatal liver toxicity from too much Paracetamol) while still achieving perfect pain relief.
Math Rule: ½ + 1 = 2 (or 0 + 1 = 2)
Potentiation happens when you mix a drug that has an active therapeutic effect with a substance that has little to zero therapeutic effect on its own. However, this seemingly useless second substance massively amplifies the power of the first drug.
- Clinical Example: Amoxicillin + Clavulanic Acid (sold together as Augmentin).
- The Problem: Many bacteria have evolved a defense shield—an enzyme called beta-lactamase, which chemically breaks open and destroys the antibiotic Amoxicillin before it can kill the bacteria.
- The Solution: Clavulanic acid has almost zero ability to kill bacteria on its own. However, it is an expert "suicide inhibitor" that irreversibly binds to and destroys the bacteria's beta-lactamase shield.
- The Result: By giving Clavulanic acid (which does nothing alone), it completely opens the door for Amoxicillin to rush in, bind to penicillin-binding proteins (PBPs), and kill the bacteria. The antibiotic activity is highly potentiated.
Math Rule: 1 + 1 = 3 (or more)
Synergism is a stronger, more powerful interaction than potentiation. The total effect produced by combining two active drugs is much greater than the simple sum of their individual effects. They team up to create a massive, multiplied response, often by blocking sequential steps in a single metabolic pathway.
- Clinical Example: Sulfamethoxazole + Trimethoprim (sold as Co-trimoxazole).
- Expanded Mechanism: Bacteria must synthesize their own folic acid from scratch to create DNA and survive. This requires a multi-step assembly line.
Step 1: Sulfamethoxazole acts as a competitive inhibitor of the enzyme dihydropteroate synthase.
Step 2: Trimethoprim blocks the very next enzyme in the chain, dihydrofolate reductase (DHFR). - The Result: Used alone, each drug merely slows the bacteria down (they are bacteriostatic). But used together, they completely and sequentially shut down the folic acid factory, turning a weak antibacterial effect into a highly lethal, synergistic bacterial wipeout (bactericidal effect).
Math Rule: 1 + 1 < 2 (or 1 + 1 = 0)
Antagonism occurs when one drug actively opposes, reduces, or completely blocks the effect of another drug. The combined effect is actually less than expected.
- Clinical Example: Opioids (Morphine/Heroin) + Naloxone.
- Expanded Mechanism: If a patient takes an opioid, it powerfully binds to mu-opioid receptors in the brain to slow breathing, induce euphoria, and stop pain. If the dose is too high, breathing stops entirely (fatal overdose).
- The Result: If you give Naloxone intravenously, it acts as a perfect competitive antagonist with a much higher affinity for the receptor than the opioid. It aggressively rips the opioid off the receptor and blocks it without activating it. The effect of the opioid drops to zero instantly, waking the patient up and saving them from an overdose.
Part 2: The Five Mechanisms of Drug Antagonism
When one drug blocks another (Antagonism), it can happen in five entirely different biological ways. Understanding exactly how the blockade happens—whether in the blood, in the liver, or at the receptor level—is critical for pharmacology exams.
1. Chemical Antagonism
This is the most basic physical interaction. Chemical antagonism occurs when two substances react directly with each other in the body fluids (like the blood, the stomach, or the gut lumen) before they even reach a cell receptor. They physically bind together to form an inactive complex, leading to the inactivation of one or both substances.
Real-World Examples of Chemical Antagonism
- Chelating Agents (e.g., Dimercaprol or EDTA): If a patient has heavy metal poisoning (like swallowing lead, arsenic, or mercury), doctors inject a chelating agent. This drug acts like a chemical claw, physically grabbing the heavy metal molecules floating in the blood and forming highly stable, inactive pairings (chelates) that are water-soluble and safely peed out by the kidneys.
- Antacids (e.g., Aluminium hydroxide): If a patient has severe heartburn or a peptic ulcer, they take an antacid. The basic aluminium hydroxide directly collides with the acidic gastric hydrochloric acid (HCl) in the stomach. They undergo a simple acid-base chemical neutralization reaction, turning into harmless salt and water, instantly neutralizing the stomach acidity.
- Heparin + Protamine Sulfate (Extra Example): Heparin is a highly negatively charged blood thinner. If a patient is bleeding out from too much Heparin, doctors give Protamine Sulfate, a highly positively charged molecule. The positive and negative charges instantly bind together chemically, neutralizing the blood thinner.
2. Pharmacokinetic Antagonism
Pharmacokinetics is the study of how the body handles a drug (Absorption, Distribution, Metabolism, Excretion - ADME). Pharmacokinetic antagonism occurs when one substance physically reduces the effective concentration of another drug by messing with its journey through the body.
This happens in three main ways:
- Reduced Absorption:
- Example: Tetracyclines (an antibiotic) + Iron salts (or milk/calcium).
- Mechanism: If a patient takes iron or calcium supplements at the same time as their tetracycline pill, the heavy metals physically bind to the antibiotic in the stomach and intestines to form an insoluble chelate. This makes the antibiotic molecule too heavy, rigid, and bulky to pass through the lipid gut wall into the blood. It totally prevents absorption, and the life-saving drug is lost in the feces.
- Enhanced Elimination via Increased Metabolism:
- Example: Phenobarbitone + Warfarin.
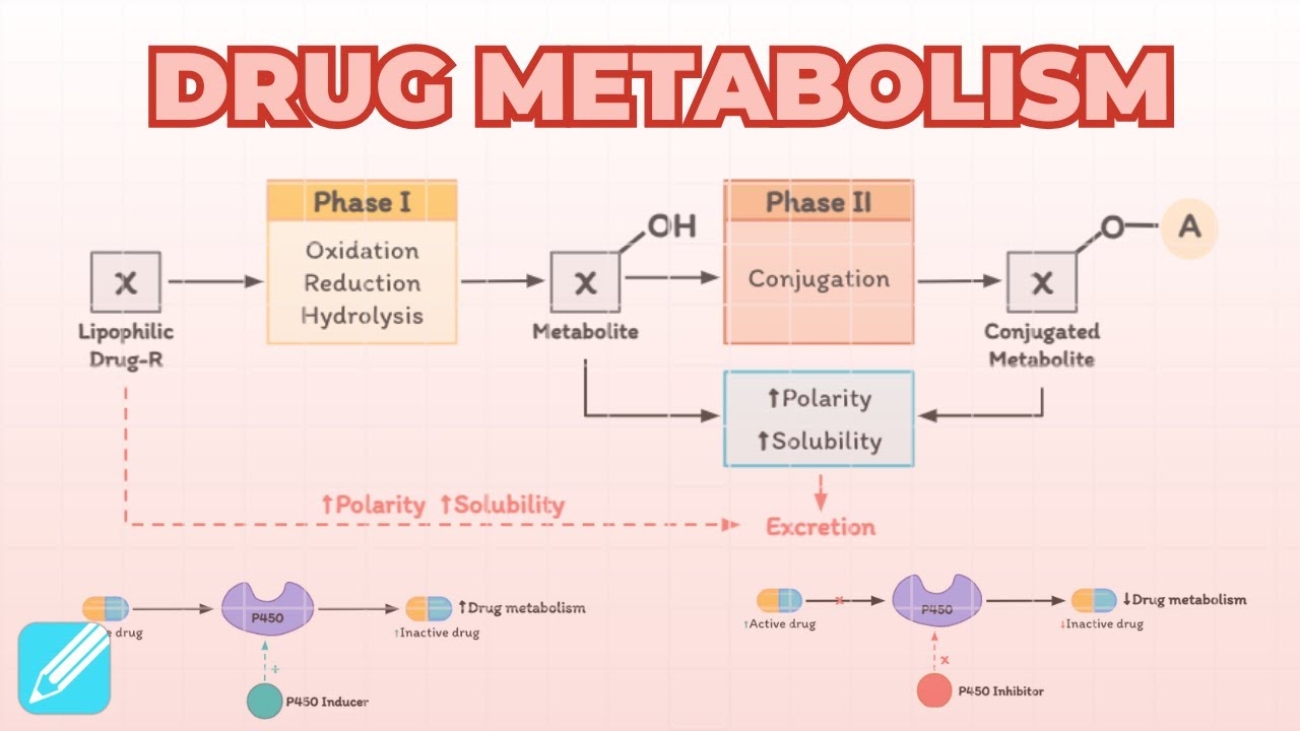
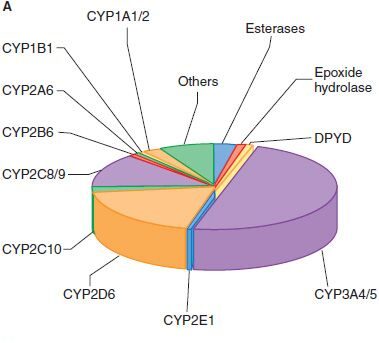
- Mechanism: Warfarin is a dangerous blood thinner metabolized by the liver's Cytochrome P450 (CYP450) enzyme system. Phenobarbitone is a drug that causes "enzyme induction"—it forces the liver's DNA to build massive amounts of brand new CYP450 drug-destroying enzymes. If you take them together, the newly boosted liver chews up and destroys the Warfarin far too quickly, severely lowering its concentration in the blood and putting the patient at severe risk for fatal blood clots.
- Enhanced Elimination via Increased Excretion (Ion Trapping):
- Example: Sodium bicarbonate + Weak acids (like Aspirin/Salicylates).
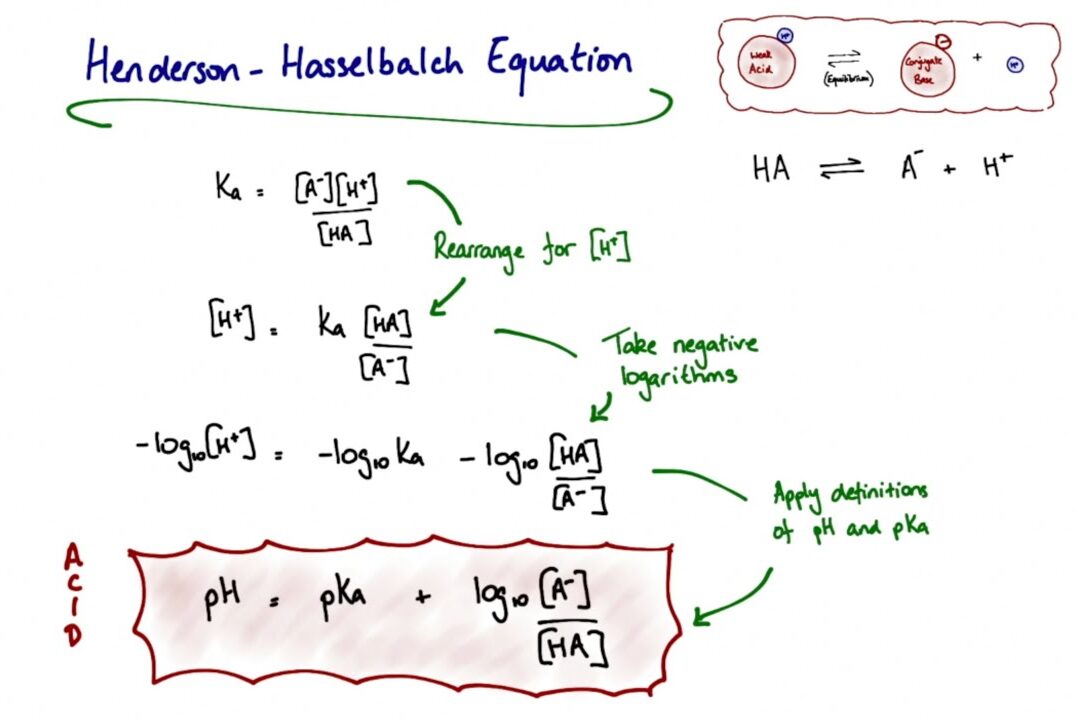
- Mechanism: In cases of an Aspirin overdose, the drug freely passes back from the kidney tubules into the blood. To stop this, doctors give IV sodium bicarbonate. This alkalinizes the urine (makes it basic, pH > 7.5). Because Aspirin is a weak acid, it donates a proton in the basic urine and becomes ionized (electrically charged). Charged molecules cannot cross lipid cell membranes. Therefore, the Aspirin gets chemically "trapped" in the basic urine and cannot be reabsorbed by the kidneys. The body excretes it incredibly fast.
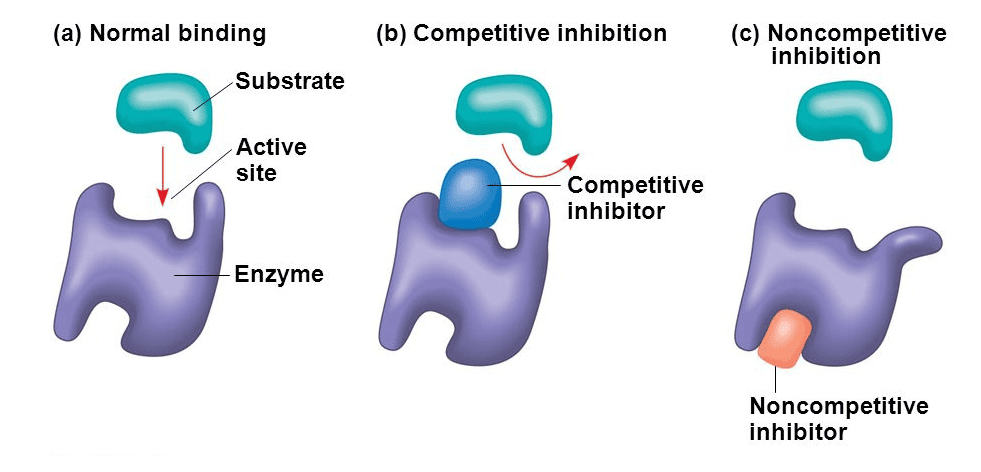
3. Competitive Antagonism (Receptor Blockade)
In this scenario, we enter the microscopic world of cellular receptors. Competitive antagonism means the antagonist and the agonist are fighting for the exact same seat (the active receptor binding site) on the cell. By physically occupying the receptor, the antagonist blocks the agonist from sitting down and producing its effect.
There are two types of competitive antagonism based on how strongly they hold onto the receptor:
A. Reversible (Surmountable) Competitive Antagonism
- Mechanism: The antagonist binds to the receptor using weak, non-covalent bonds (like hydrogen, van der Waals, or ionic bonds). Because the grip is weak, it frequently lets go and readily dissociates from the receptor.
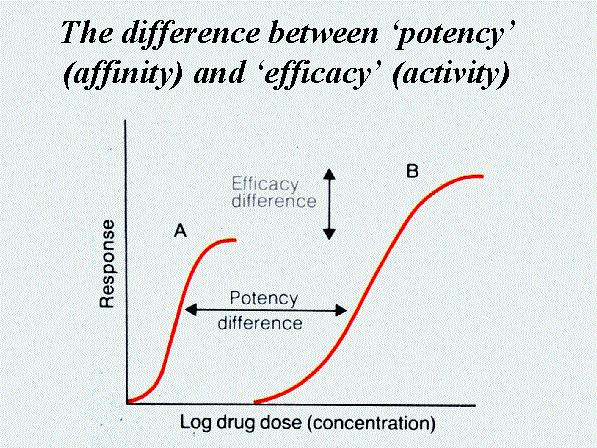
- The Key Feature: It is surmountable. This means if you flood the area with a massive concentration of the agonist, the agonist will mathematically outnumber the antagonist and win the game of musical chairs, displacing the antagonist and completely overcoming the blockade. (On a graph, this causes a parallel shift of the dose-response curve to the right).
- Clinical Example: Propranolol. This is a reversible beta-blocker drug. It competitively sits on the beta-receptors of the heart, temporarily blocking the body's natural catecholamines (adrenaline and noradrenaline) from speeding up the heart rate. A massive surge of adrenaline can overcome it.
B. Irreversible (Non-surmountable) Competitive Antagonism
- Mechanism: The antagonist binds to the receptor using incredibly strong covalent bonds, or with extremely high affinity that essentially never lets go. It is like using industrial superglue. It permanently inactivates that specific receptor.
- The Key Feature: It is non-surmountable. Because the receptor is permanently broken and occupied, increasing the agonist concentration does absolutely nothing. The maximum possible response of the tissue is severely reduced. The response cannot be fully restored until the cell takes days to manufacture brand new receptors from scratch via DNA transcription. (On a graph, this causes a downward shift of the maximum efficacy curve).
- Clinical Example: Phenoxybenzamine. This is an alpha-adrenergic blocker used in tumors like pheochromocytoma. It irreversibly binds to alpha-receptors on blood vessels, permanently preventing noradrenaline from causing deadly vasoconstriction.
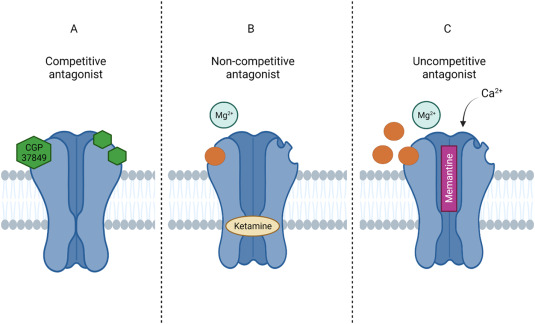
4. Non-Competitive Antagonism (Allosteric Blockade)
This is a highly clever form of antagonism. The antagonist does not fight for the same front-row seat. Instead, a non-competitive antagonist binds to a completely different, separate location on the receptor called an allosteric site.
- Mechanism: By sitting on this separate allosteric site, the antagonist forces the entire 3-dimensional protein structure of the receptor to change its physical shape (conformational change). Because the overall shape is changed, the main active binding site is ruined, and the agonist can no longer fit into it, or if it does fit, it can no longer trigger a signal.
- The Key Feature: It is entirely insurmountable. You can add a million molecules of the agonist, but it won't matter because the main doorway is physically deformed and non-functional. (Note: The antagonist itself can be held by reversible or irreversible bonds to the allosteric site, but the effect on the agonist is always insurmountable).
- Clinical Example 1: Ketamine. Acts as a non-competitive antagonist at NMDA receptors in the brain, changing their shape and completely blocking excitatory glutamate neurotransmission (creating profound dissociative anesthesia).
- Clinical Example 2: Maraviroc. A powerful anti-HIV drug. It binds to a side-site on human white blood cell CCR5 receptors. This alters the shape of the main receptor so severely that the HIV virus cannot attach its gp120 protein to the cell to enter it.
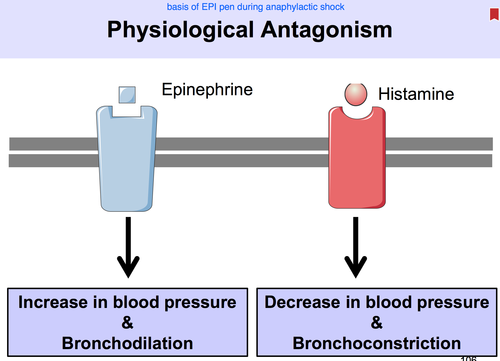
5. Physiological Antagonism
This is a biological tug-of-war at the organ level. Physiological antagonism occurs when two completely different drugs (or natural hormones) act on completely different receptors and utilize entirely different intracellular pathways to produce exact opposite physiological effects. There is absolutely no direct competition at the same receptor.
Clinical Scenarios of Physiological Antagonism
- Insulin vs. Glucagon (Blood Sugar Control):
- Insulin binds to tyrosine-kinase insulin receptors, commanding the body to lower blood glucose by promoting cellular sugar uptake, glycolysis, and glycogen storage.
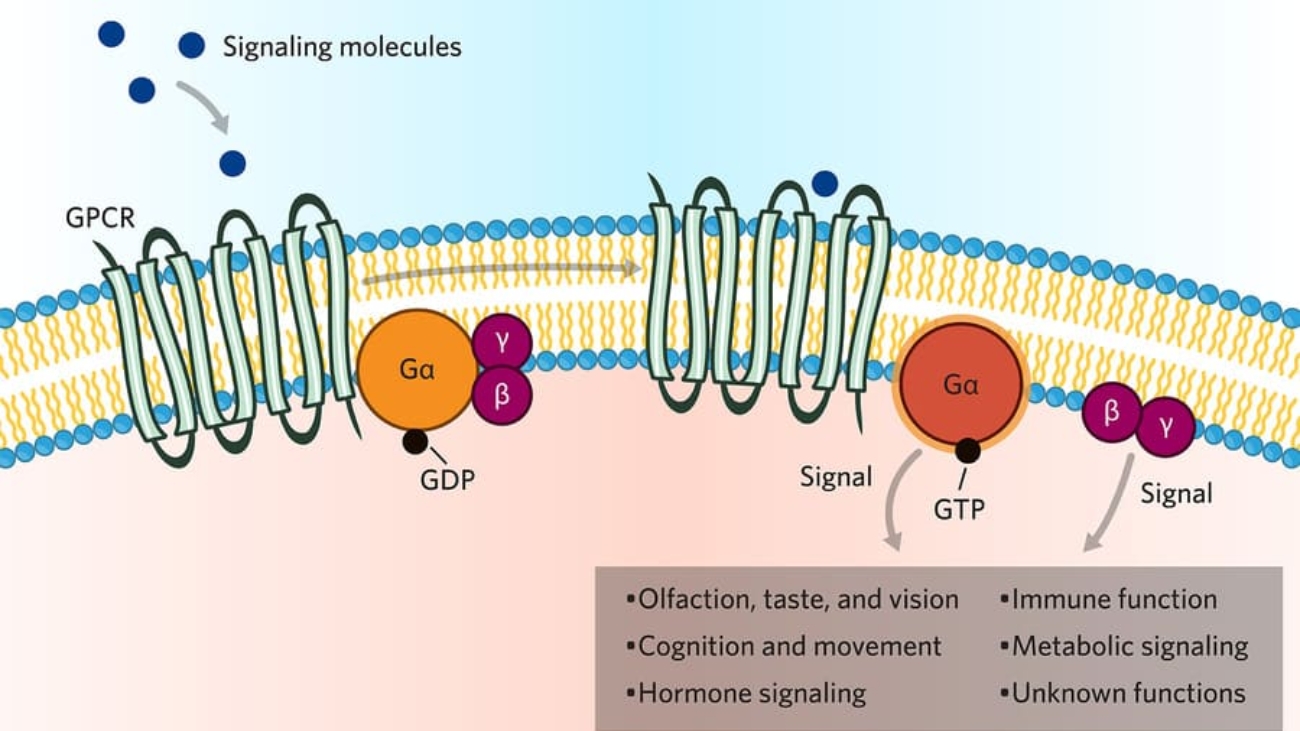
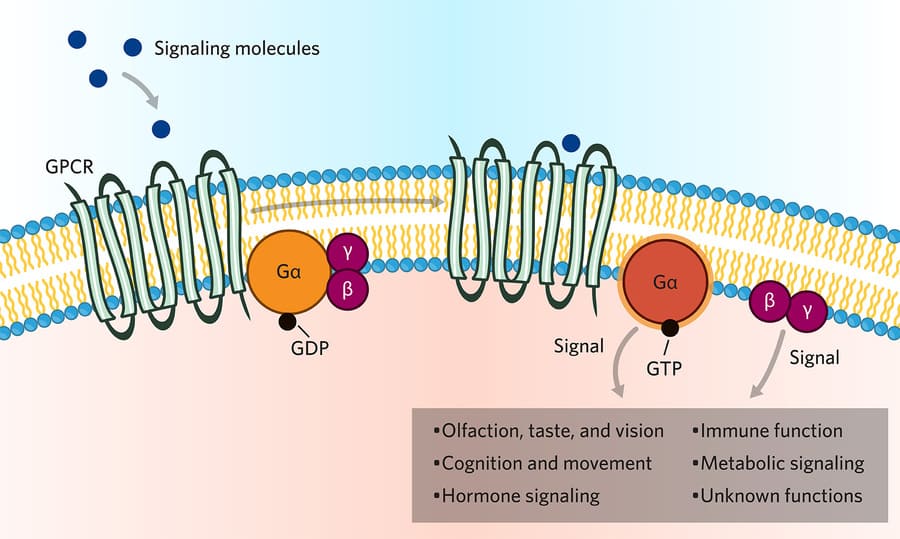
- Glucagon binds to G-protein coupled glucagon receptors, commanding the body to raise blood glucose by stimulating the liver to break down glycogen and create new sugar (gluconeogenesis). They fight each other constantly via separate pathways to maintain perfect homeostasis.
- Histamine vs. Adrenaline (The EpiPen Mechanism for Anaphylaxis):
- If you are allergic to a bee sting, your mast cells release massive amounts of Histamine. Histamine binds to H1 receptors, causing deadly bronchospasm (throat closing via smooth muscle contraction) and severe vasodilation/hypotension (crashing blood pressure).
- To save your life, you inject Adrenaline (Epinephrine). Adrenaline ignores the H1 receptors entirely. Instead, it binds to Beta-2 adrenergic receptors in the lungs (causing powerful bronchodilation/opening the throat) and Alpha-1 adrenergic receptors in the vessels (causing severe vasoconstriction/raising blood pressure). It directly and physiologically counteracts the deadly effects of histamine to save the patient.

Part 3: Loss of Drug Effect over Time
A major challenge in clinical medicine is that the therapeutic effect of a drug may diminish with continuous or repeated administration. The body learns to adapt to the drug, making it weaker. We use four distinct terms to describe this phenomenon based on how fast it happens and what is causing it.
- Tachyphylaxis (Desensitization): This is incredibly fast and rate-dependent. It is a rapid, acute loss of drug effect that occurs within mere minutes or hours of administration. Crucially, simply increasing the dose will NOT restore the effect.
- Example: Repeated doses of ephedrine or amphetamines (drugs that act indirectly by squeezing nerve terminals to release stored noradrenaline). If you give it continuously, you squeeze the nerve entirely dry. The noradrenaline stores are depleted, and subsequent doses produce progressively smaller, useless responses until the nerve has time to manufacture more neurotransmitter.
- Tolerance: This is much slower and dose-dependent. It is a gradual decrease in physiological responsiveness developing over days, weeks, or months of taking a medication. The effect can usually be restored by giving a larger dose.
- Example: Chronic use of powerful painkillers like opioids (morphine). Over weeks of use, the patient's body adapts (often by uncoupling the opioid receptors from their internal G-proteins), meaning they require higher and higher doses just to achieve the exact same baseline analgesic (pain-relieving) effect.
- Refractoriness: A stubborn, absolute state where a drug that was previously highly effective simply no longer produces any therapeutic response at all, regardless of massive dose increases.
- Example: Patients taking Nitrates for chest pain (angina). After prolonged continuous use without a break, the blood vessels deplete critical sulfhydryl (-SH) groups needed to process the drug, and refuse to respond to the nitrates entirely. (Doctors solve this by enforcing an 8-to-12-hour "nitrate-free" period every night to let the body regenerate its enzymes and reset).
- Drug Resistance: This strictly refers to the loss of effectiveness of antimicrobial (antibiotic), antiviral, or anticancer drugs. It is not the human body adapting; it is due to rapid genetic, adaptive changes (mutations or horizontal gene transfer) in the target foreign organism or the mutated tumor itself.
- Example: Bacterial resistance to antibiotics. A classic threat is MRSA (methicillin-resistant Staphylococcus aureus), a bacterium that mutated to build armor (altering its penicillin-binding proteins) against penicillin-style drugs, making standard antibiotics useless.
Part 4: The 6 Biological Mechanisms Causing Loss of Drug Effect
Why does tolerance or tachyphylaxis happen at the microscopic cellular level? The body treats drugs as foreign disruptions and employs six distinct defense mechanisms to fight off constant drug exposure and return to homeostasis.
Receptor Desensitization / Uncoupling
The physical receptor on the cell undergoes rapid functional modifications (often via phosphorylation by specialized kinases). The drug can still successfully bind to the receptor's surface, but the internal wiring is unplugged—signal transduction to the G-protein is completely impaired.
- Example: Beta-adrenergic receptor desensitization. If an asthmatic patient overuses their salbutamol inhaler (a beta-agonist), the lung receptors become exhausted and structurally uncoupled. They stop sending the internal cAMP signal to open the airways, making the inhaler useless during an acute attack.
Receptor Downregulation
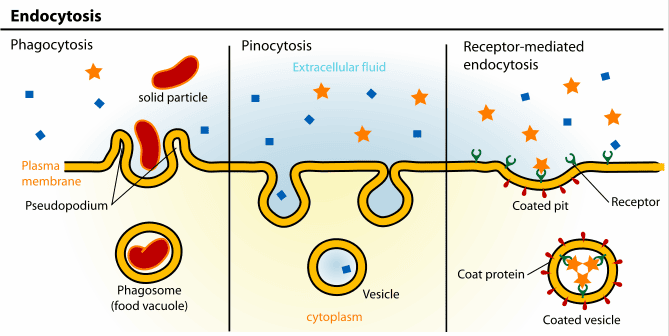
The cell realizes it is being dangerously overstimulated. To protect itself from burnout, prolonged drug exposure causes the cell to actively pull receptors inside the cell membrane (endocytosis) and destroy them in lysosomes, massively reducing the total number of receptors available on the cell surface.
- Example: Continuous exposure to high blood insulin (as seen in obesity and early Type 2 Diabetes). The cells downregulate their insulin receptors to avoid absorbing toxic levels of sugar. Once the drug/hormone is withdrawn, recovery to baseline receptor levels is very slow because the cell must synthesize new proteins.
Depletion of Endogenous Chemicals
Some drugs do not act directly on end-organ receptors; they act indirectly by forcing the body to dump a stored chemical mediator. Repeated, rapid drug stimulation completely depletes these essential pre-packaged mediators required for the effect.
- Example: Indirect sympathomimetics like ephedrine or amphetamines lose effectiveness rapidly (tachyphylaxis) after repeated dosing strictly due to the total depletion of the body's synaptic noradrenaline vesicles.
Pharmacokinetic Tolerance (Auto-Induction)
The liver treats drugs like poisons. Repeated drug exposure can actively induce (ramp up the DNA transcription of) liver metabolic enzymes like the Cytochrome P450 system. This leads to vastly accelerated drug breakdown, heavily reducing the drug's half-life and clinical effectiveness.
- Example: Tolerance to ethanol (alcohol) or barbiturates. A heavy drinker's liver builds massive amounts of hepatic alcohol dehydrogenase and microsomal CYP enzymes. They process the alcohol so fast that the individual requires huge amounts of liquor to feel intoxicated compared to a novice.
Homeostatic Compensation
The body constantly wants to remain at its programmed baseline (homeostasis). If a drug shifts the baseline (e.g., dropping blood pressure), the body activates robust, entirely separate counter-regulatory mechanisms to aggressively oppose the drug’s intended effect.
- Example: A patient takes long-term thiazide diuretics to lower their blood pressure by urinating out excess fluid. The kidneys panic at the loss of blood volume and stimulate the Renin-Angiotensin-Aldosterone System (RAAS), a physiological pathway designed to violently retain salt and water, actively reducing and opposing the diuretic's efficacy over time.
Efflux Pumps
The cells build biological "sump pumps" to actively spit the drug back out into the blood or gut. Cells may massively increase the genetic expression of efflux pumps or transporters to physically remove the drug, fatally reducing intracellular drug concentrations.
- Example: Chemotherapy resistance. Cancer cells are notoriously highly adaptable. They overexpress P-glycoprotein (MDR1 - Multi-Drug Resistance Protein), a heavy-duty, ATP-powered pump that actively captures toxic anticancer drugs that enter the cell and violently pumps them back out into the blood before they can reach the nucleus to kill the tumor.
Part 5: Master Glossary of Receptor Interactions
To ensure perfect clarity for your exams, here is a definitive breakdown of exactly how substances are defined based on how they interact with cellular receptors and other drugs:
| Type of Substance | Effect / Definition |
|---|---|
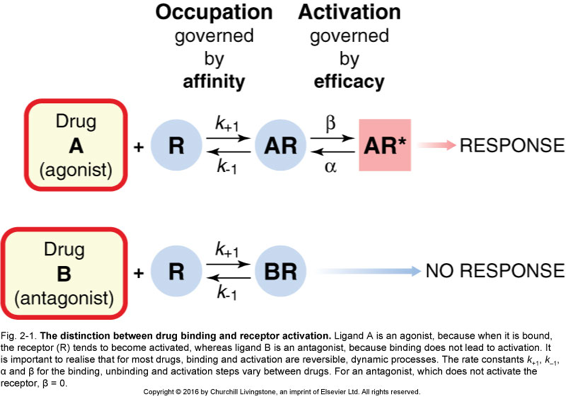
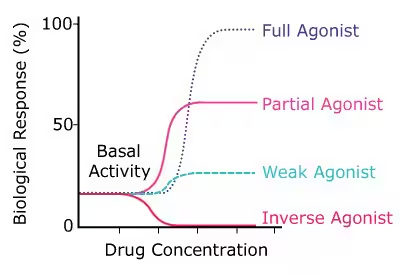
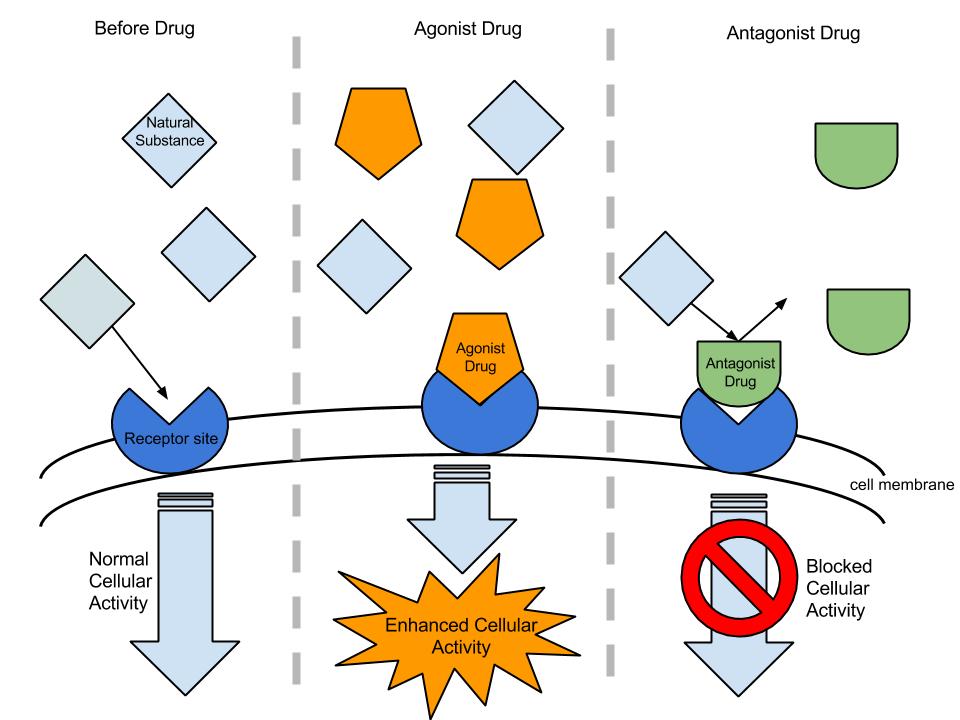
| Agonist | A key that perfectly fits the lock. It binds to a receptor, possesses high affinity, and actively turns the receptor "on" (high intrinsic activity) to produce a full physiological response. (e.g., natural Adrenaline or Morphine). |
| Antagonist | A key that fits into the lock, but cannot turn it. It binds to a receptor site (has affinity) strictly to block other agonists from entering and causing effects. It has absolutely zero intrinsic activity of its own. |
| Inverse Agonist | A highly unique substance that binds to the exact same site as the agonist, but forces the receptor to produce an effect that is the exact mathematical opposite to that of a normal agonist. (It actively lowers the baseline, constitutive activity of the receptor below normal). |
| Superagonist | A synthetic laboratory substance that produces a much greater, exaggerated maximum response from a receptor than the natural, endogenous (body-made) substance ever could (Efficacy > 100%). |
| Partial Agonist | A weak key. A substance that binds to the receptor but has only partial efficacy (intrinsic activity between 0 and 1). Even if it completely saturates and fills 100% of the receptors, it can never produce the maximum full response that a full agonist can. (It can actually act as an antagonist if a full agonist is present!). |
| Additive Interaction | Occurs when the effects of two drugs simply summate without enhancing each other. It is a straight, basic algebraic addition of effects (1 + 1 = 2). |
| Potentiation / Synergism | Exposure to one drug produces an amplified, multiplied effect on a second drug, pushing the total therapeutic power far beyond simple addition (1 + 1 = 3 or more). |