Drug Metabolism (Biotransformation)
Many students fear "Drug Metabolism" because of the heavy biochemistry and enzyme names. Do not panic. Think of drug metabolism simply as the body's waste management system. The body wants to get rid of foreign chemicals (drugs). To do this, it must change their shape and properties so they can be flushed down the drain (kidneys). This guide will break down every mechanism, enzyme, and clinical scenario so you understand the "why" behind the science.
Drug Metabolism
Drug metabolism, also known as Biotransformation, is a core pillar of Pharmacokinetics. Remember the acronym ADME: Absorption, Distribution, Metabolism, and Excretion. Metabolism bridges the gap between a drug moving through your tissues and a drug leaving your body.
The Journey of a Drug:
- Dose → Absorption → Blood Plasma (Free vs. Protein-Bound) → Distribution to Tissues/Receptors (Effect) → METABOLISM → Elimination (Renal Excretion).
Why is Metabolism Absolutely Necessary?
- The Fundamental Problem: Most drugs that enter the body are designed to be lipophilic (fat-soluble). They need to be lipophilic so they can easily diffuse through the lipophilic cell membranes in your gut to be absorbed, and cross into tissues (like the brain) to exert their effects.
- The Catch-22: The kidneys (the body's main filter) cannot efficiently excrete lipophilic drugs. When blood is filtered through the renal glomerulus, lipid-soluble drugs simply slide right back through the renal tubule membranes and are reabsorbed back into the systemic circulation. If we couldn't metabolize them, lipophilic drugs would stay in the body forever, leading to massive accumulation and fatal toxicity.
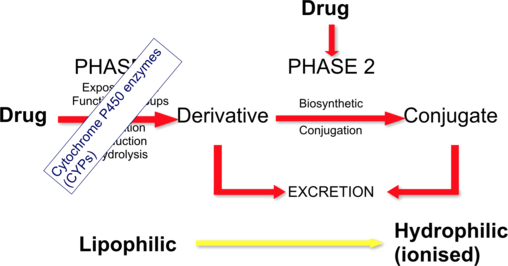
The Solution: The Definition of Metabolism
Metabolism is the process where a drug is structurally altered (biotransformed) to become more polar and hydrophilic (water-soluble) so that it can be trapped in the urine and excreted.
Analogy: Imagine trying to wash engine grease (a lipophilic drug) off your hands using only water (urine). It doesn't work; the grease clings to your skin. You need soap (metabolism) to chemically alter the grease, making it mix with water so it can be rinsed down the drain.
The Four Consequences of Drug Alteration
When the body chemically alters a drug, four different clinical outcomes can occur. Metabolism doesn't just mean destroying a drug; it means changing its pharmacological activity.
Active Drug → Inactive Metabolite
The standard detoxifying process. The drug does its job, and the liver shuts it off.
- Examples: Paracetamol, Ibuprofen, Chloramphenicol.
Active Drug → Active or Toxic Metabolite
Sometimes, the body's attempt to alter a drug creates a byproduct that still has a therapeutic effect, or worse, is highly toxic.
Inactive Prodrug → Active Drug
A Prodrug is a drug administered in an inactive form. It relies entirely on the body's metabolism to activate it. We use prodrugs to improve absorption or bypass harsh stomach acid.
Unexcretable (Lipophilic) → Excretable (Hydrophilic)
The structural conversion that allows final renal clearance and removal from the body.
Consequence Tables
| Original Active Drug | Active/Toxic Metabolite Formed (Outcome 2) |
|---|---|
| Allopurinol (Gout medication) | Alloxanthine (Also lowers uric acid) |
| Digitoxin | Digoxin (Active heart medication) |
| Morphine | Morphine-6-glucuronide (Highly active painkiller) |
| Chloral hydrate | Trichloroethanol |
| Inactive Prodrug | Active Metabolite (The functional drug) (Outcome 3) |
|---|---|
| Levodopa (Crosses Blood-Brain Barrier) | Dopamine (Treats Parkinson's Disease) |
| Sulindac | Sulfide metabolite |
| Prednisone | Prednisolone (Active anti-inflammatory) |
Where Does Drug Metabolism Occur?
While enzymes capable of biotransformation exist in almost every tissue (gut, lung, kidney, skin, placenta), the LIVER is the undisputed chief organ for drug metabolism. Nearly 90% of all drug metabolism happens here.
Why the Liver?
- Blood Supply: The liver receives massive blood flow, specifically from the portal vein, which brings blood directly from the digestive tract.
- Enzyme Concentration: Liver cells (Hepatocytes) contain the body's full complement of metabolizing enzymes in their smooth endoplasmic reticulum (ER), cytosol, and mitochondria.
The First-Pass Effect
When you swallow a pill (PO - Per Os), it is absorbed in the intestines and goes into the portal vein. The portal vein goes straight to the liver before the drug reaches the rest of the body. The liver enzymes immediately metabolize a large portion of the drug. This "first-pass" can drastically reduce the amount of active drug that makes it to systemic circulation (bioavailability). If a drug has massive first-pass metabolism, it must be given via IV, sublingually, or transdermally to bypass the liver initially.
Levels of Metabolism by Organ:
- High: Liver
- Medium: Lung, Kidney, Intestine
- Low: Skin, Testes, Placenta, Adrenals
- Very Low: Nervous System
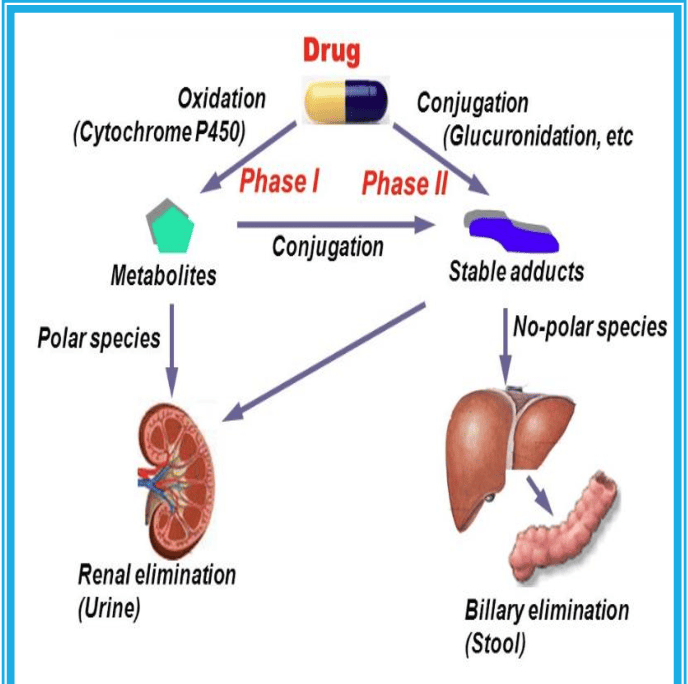
Reactions of Drug Metabolism: Phase I & Phase II
To turn a stubborn, lipophilic drug into a water-soluble waste product, the liver uses a two-step process: Phase I and Phase II. (Note: Not all drugs go through both; some skip Phase I, some skip Phase II, and some go in reverse, but the standard sequence is I → II).
Phase I Reactions: Modification (Functionalization)
Goal: To modify the drug by unmasking or adding a small, polar "chemical hook" (like an -OH, -NH2, or -COOH group). This makes the drug slightly more water-soluble, but more importantly, it provides a handle for Phase II enzymes to grab onto.
Types of Phase I Reactions:
- Oxidation: The most common. Involves the addition of oxygen or removal of hydrogen (e.g., converting a C-H bond to a C-OH bond: Hydroxylation, Dealkylation).
- Reduction: Addition of hydrogen.
- Hydrolysis: Breaking bonds using water.
The Enzymes of Phase I
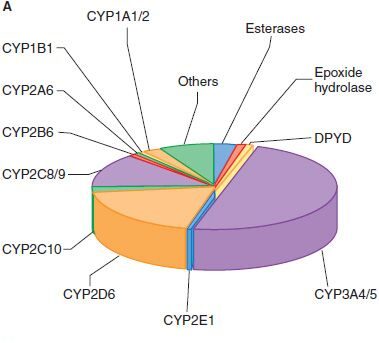
Phase 1 is dominated by the Cytochrome P450 (CYP450) superfamily of enzymes, responsible for >95% of oxidative metabolism. They are located in the microsomes of the smooth endoplasmic reticulum (hence called microsomal monooxygenase enzymes).
Understanding CYP450 Nomenclature:
There are at least 18 different forms in humans. Take the most important one: CYP3A4 (which metabolizes ~50% of all drugs alone).
- CYP = Cytochrome P450
- 3 = Family (Families 1, 2, and 3 handle most drug metabolism)
- A = Subfamily
- 4 = Specific individual enzyme gene
Overall, 60% of all drugs are metabolized primarily by the CYP450 family.
Non-CYP Enzymes in Phase I (<5% of metabolism):
- Monoamine Oxidase (MAO): Found in mitochondria. Oxidizes endogenous neurotransmitters (dopamine, serotonin, epinephrine) and drugs related to them.
- Alcohol & Aldehyde Dehydrogenase: Found in liver cytosol. Responsible for breaking down ethanol (alcohol).
- Xanthine Oxidase (XO): Converts hypoxanthine to xanthine, and then to uric acid. Target for gout drugs (Theophylline, 6-mercaptopurine).
- Esterases: Hydrolyze endogenous substances (e.g., Acetylcholinesterase breaks down acetylcholine).
Phase II Reactions: Conjugation
Goal: If Phase I added a "hitch" to the drug, Phase II attaches a massive, heavy, highly polar "trailer" to that hitch. This process is called Conjugation.
Phase II enzymes attach large, endogenous, water-soluble molecules to the -OH, -NH2, or -SH functional groups created in Phase I. This effectively inactivates the drug and makes it highly lipid-insoluble, guaranteeing its rapid excretion in urine or bile.
The 6 Types of Phase II Conjugation Reactions
These require specific transferase enzymes to link the endogenous compound to the drug.
- Enzyme: UDP-glucuronosyltransferase.
- Details: The most common Phase II reaction. It is highly inducible.
- Examples: Digoxin, Morphine, Paracetamol, Bilirubin.
- Clinical Scenario (Neonates): Newborns have very low activity of UDP-glucuronosyltransferase. Bilirubin accumulation causes Neonatal Jaundice. Lack of conjugation of chloramphenicol causes the fatal "Gray Baby Syndrome."
- Enzyme: Acetyltransferase.
- Examples: Isoniazid (Anti-TB drug), Dapsone, Hydralazine, Procainamide.
- Clinical Scenario (Genetics): Humans are genetically divided into "Fast" and "Slow" acetylators. Slow acetylators taking Hydralazine or Procainamide can develop Drug-Induced Systemic Lupus Erythematosus (SLE).
- Enzyme: Glutathione transferase.
- Details: Glutathione is the body's primary antioxidant. This pathway neutralizes free radicals and highly reactive, toxic metabolites.
- Examples: Methyldopa, Paracetamol.
- Enzyme: Methyltransferase.
- Example: Inactivation of Histamine.
- Enzyme: Sulphate transferase.
- Examples: Almost all steroid hormones, Salbutamol (asthma inhaler), Paracetamol.
- Details: Less common, generally pairs with benzoic acid derivatives.
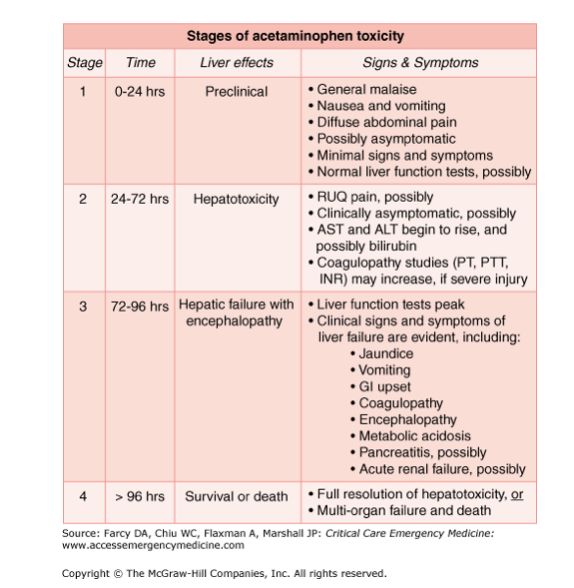
Paracetamol (Acetaminophen) Toxicity Pathway
This is a classic, heavily tested scenario that proves why Phase I and Phase II balance is critical for survival.
- Normal Doses: ~95% of paracetamol skips Phase I entirely. It goes straight to Phase II, where it safely undergoes Glucuronidation and Sulfation to form non-toxic metabolites excreted in urine.
- The Danger (Phase I): The remaining ~5% goes through Phase I via CYP450 2E1. This oxidation creates a highly reactive, highly toxic free radical metabolite called NAPQI (N-acetyl-p-benzo-quinone imine).
- The Savior (Phase II): Normally, Phase II Glutathione conjugation immediately binds to NAPQI, neutralizing it into harmless mercapturic acid.
- The Overdose Scenario: In an overdose, the glucuronidation and sulfation pathways get saturated (full). The body pushes the massive excess of paracetamol down the CYP450 2E1 pathway, generating massive amounts of toxic NAPQI. The liver's supply of Glutathione is rapidly depleted. Unbound NAPQI begins binding covalently to hepatic cell proteins, causing severe Hepatotoxicity (liver death).
- The Antidote: We administer NAC (N-acetylcysteine), which acts as a substitute for depleted glutathione to bind and neutralize the toxic NAPQI.
Factors Affecting Drug Metabolism
Drug metabolism is an enzymatic process. Therefore, anything that affects enzymes affects metabolism. This leads to massive inter-individual variability (why a dose that cures one patient might poison another).
A. Environmental Factors: Enzyme Induction vs. Inhibition
Exposure to certain exogenous compounds (other drugs, food, environmental pollutants, smoke) can modulate enzyme activity.
1. Enzyme Induction (Speeding up)
- Mechanism: Exposure to an inducer stimulates the DNA to synthesize more CYP450 enzymes. The metabolic capacity increases, so the drug is metabolized and cleared much faster.
- Consequences of Induction:
- Increased rate of metabolism.
- Decreased plasma concentration of the drug.
- Reduced bioavailability and Reduced efficacy (the drug stops working).
- Exception: If the drug is a prodrug or has a toxic metabolite, induction leads to increased toxicity.
- Therapeutic Implication: Dosing rates must be increased to maintain effective blood levels. Be careful: it takes days to weeks for induction to fully occur and wear off.
- Classic General Inducers: Anticonvulsants (Phenobarbital, Phenytoin, Carbamazepine), Antibiotics (Rifampin), Chronic Alcohol use, St. John's Wort. Cigarette smoking specifically induces CYP1A2 (smokers require higher doses of Theophylline).
2. Enzyme Inhibition (Slowing down)
- Mechanism: Inhibitors block the enzyme from working. This can occur rapidly with no warning. Types of inhibition include:
- Competition: A high-affinity drug hogs the active site, slowing metabolism of a lower-affinity drug.
- Irreversible Inactivation: The drug forms a complex with the heme iron of CYP450 (e.g., Cimetidine, Ketoconazole) or destroys the heme group entirely (Secobarbital).
- Depletion of Cofactors: E.g., running out of NADH2 for Phase II.
- Consequences of Inhibition:
- Increase in plasma concentration of the parent drug.
- Reduction in metabolite concentration.
- Exaggerated, prolonged pharmacological effects.
- High likelihood of drug-induced toxicity.
- Classic General Inhibitors: Anti-ulcer meds (Cimetidine, Omeprazole), Antimicrobials (Chloramphenicol, Macrolides, Ritonavir, Ketoconazole, Quinolones/Ciprofloxacin), Acute Alcohol ingestion, Grapefruit Juice (potent inhibitor of CYP3A4).
B. Disease Factors
Since the liver is the primary metabolic factory, Liver Disease (Cirrhosis, Alcoholic liver disease, Jaundice, Hepatic Carcinoma) severely impairs metabolism. There is less functional liver mass and decreased enzyme activity. This reduces the first-pass effect, potentially increasing bioavailability by 2-4x, leading to exaggerated responses and severe adverse effects unless drug doses are heavily reduced.
C. Age and Sex
- Newborns and Infants: Metabolize drugs slowly because their liver enzyme systems are immature and underdeveloped.
- Adolescents/Adults: Full metabolic maturity appears in the second decade of life.
- Elderly: Experience a slow decline in metabolic function associated with aging (decreased liver mass and hepatic blood flow).
D. Genetic Variation (Polymorphism)
Genetic Polymorphism refers to the existence of multiple forms of a DNA sequence at a specific locus within a population. It creates distinct subgroups of people who differ drastically in their ability to perform biotransformation.
Changes in a single allele (Single Nucleotide Polymorphisms or SNPs) dictate the phenotype (observable physical/biochemical function) of the enzyme. Mutations can cause decreased, increased, or completely absent enzyme activity.
The Four Metabolic Phenotypes:
- Carry 2 defective alleles (e.g., gene deletions or mutations resulting in no functional enzyme).
- Result: Active drugs build up (high toxicity risk). Prodrugs fail to activate (zero efficacy).
- Heterozygous (carry one normal wild-type allele and one defective allele).
- Slower metabolism than normal, but not absent.
- Carry wild-type (normal) alleles.
- They encode normal enzyme function. This is the baseline population.
- Carry genetic duplications (two or more copies of an amplified gene).
- Result: Unusually high enzyme activity. Active drugs are cleared so fast they have no therapeutic effect. Prodrugs are activated so fast they can cause sudden toxicity.
Genetics vs. Drug Interactions
The Codeine Scenario: Codeine is an inactive prodrug. It must be metabolized (oxidized) by the enzyme CYP2D6 into Morphine to relieve pain.
- In Poor Metabolizers (PMs), codeine is never converted to morphine. They receive no pain relief.
- In Ultra-Rapid Metabolizers (UMs), codeine turns into morphine instantly, risking fatal respiratory depression.
Interplay of Genetics and Inducers/Inhibitors:
- Inhibitors affect EMs more than PMs: If you give an inhibitor to an Extensive (Normal) Metabolizer, their metabolism crashes, and you see a huge change. If you give an inhibitor to a Poor Metabolizer, they already had zero enzyme activity, so nothing changes.
- Inducers affect PMs more than EMs: Inducing an enzyme in someone who naturally metabolizes poorly causes a massive, highly noticeable relative jump in enzyme activity compared to inducing someone already functioning at maximum normal capacity.