Cholesterol Metabolism
Cholesterol often gets a bad rap due to its association with heart disease, but it's crucial to understand that it is an essential molecule for life. Our bodies need cholesterol to function properly. The problem arises when its levels are imbalanced or when it's handled improperly within the body.
Importance and Physiological Functions of Cholesterol
Cholesterol is a lipid belonging to the steroid family. Its unique amphipathic structure (a polar hydroxyl group and a nonpolar steroid ring system and hydrocarbon tail) allows it to insert into cell membranes, giving it critical structural and signaling roles.
- Essential Component of Cell Membranes:
- Cholesterol is a major constituent of virtually all animal cell membranes.
- It modulates membrane fluidity, permeability, and stability, acting as a "buffer": at high temperatures, it stiffens the membrane, while at low temperatures, it prevents rigidity.
- It is particularly abundant in myelin sheaths, enhancing nerve signal transmission.
- Precursor for Steroid Hormones:
- Cholesterol is the obligate precursor for all five major classes of steroid hormones: Glucocorticoids (e.g., Cortisol), Mineralocorticoids (e.g., Aldosterone), Androgens (e.g., Testosterone), Estrogens (e.g., Estradiol), and Progestogens (e.g., Progesterone).
- Precursor for Bile Acids (and Bile Salts):
- In the liver, cholesterol is converted into primary bile acids.
- Bile acids emulsify dietary fats in the small intestine, facilitating their absorption. This is the primary way the body eliminates excess cholesterol.
- Precursor for Vitamin D Synthesis:
- 7-Dehydrocholesterol, a precursor in the cholesterol synthesis pathway, is converted to pre-vitamin D3 in the skin upon exposure to UV light.
- This is then converted to the active hormone, calcitriol, essential for calcium homeostasis.
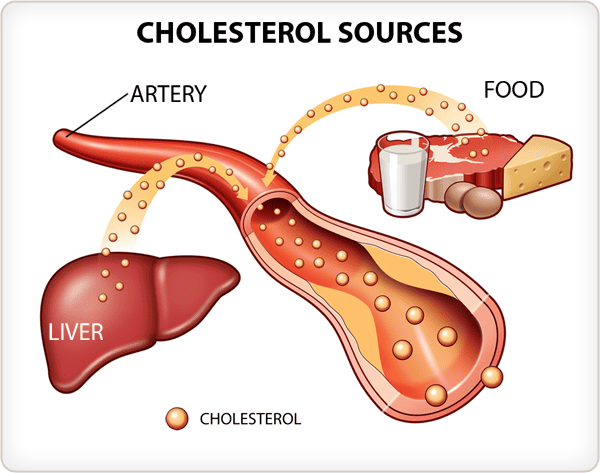
Sources of Cholesterol
The body acquires cholesterol from two main sources:
- Endogenous Synthesis (De Novo Synthesis):
- The vast majority of cholesterol (about 80%) is synthesized internally, primarily in the liver (~50% of total synthesis), but also in the intestine, adrenal cortex, and reproductive organs.
- Dietary Intake (Exogenous Cholesterol):
- Cholesterol is consumed in the diet, found exclusively in animal products (meat, eggs, dairy). Plant foods do not contain cholesterol.
- The amount absorbed can vary significantly among individuals.
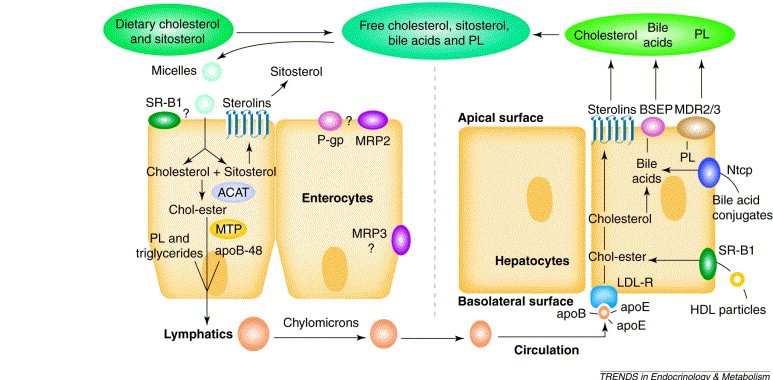
Absorption of Dietary Cholesterol
The process of dietary cholesterol absorption primarily occurs in the small intestine:
- Emulsification: Dietary cholesterol esters are emulsified by bile salts into smaller micelles.
- Hydrolysis: Cholesterol Esters (CE) are hydrolyzed into free cholesterol (FC) by pancreatic cholesterol esterase. Only free cholesterol can be absorbed.
- Micelle Formation: Free cholesterol and other digested lipids become incorporated into mixed micelles with bile salts.
- Uptake by Enterocytes:
- Mixed micelles diffuse to the brush border of the intestinal enterocytes.
- The primary transporter responsible for cholesterol uptake is the Niemann-Pick C1-Like 1 (NPC1L1) protein. This transporter is the target of the drug ezetimibe.
- Intracellular Processing and Re-esterification:
- Once inside the enterocyte, free cholesterol can be either effluxed back into the lumen via ABC G5/G8 transporters or re-esterified to cholesterol esters by the enzyme Acyl-CoA Cholesterol Acyltransferase 2 (ACAT2).
- Chylomicron Assembly and Secretion:
- The newly formed cholesterol esters and re-formed triacylglycerols are packaged with apolipoproteins (primarily apoB-48) into large lipoprotein particles called chylomicrons.
- Chylomicrons are then released into the lymphatic system, which eventually drains into the bloodstream.
Summary
So, to summarize, cholesterol is a vital molecule for cell structure, hormones, bile acids, and Vitamin D. We get it from both our diet and internal synthesis. Dietary cholesterol is absorbed in the small intestine via NPC1L1, processed, and then packaged into chylomicrons for transport.
Pathways of Cholesterol Synthesis (De Novo Synthesis)
Cholesterol is an indispensable molecule, and while we obtain some from our diet, the human body possesses the remarkable ability to synthesize nearly all the cholesterol it requires through a complex process known as de novo synthesis. This internal production ensures a constant supply for vital cellular functions.
Sites of Synthesis:
While virtually all nucleated cells can synthesize cholesterol, certain tissues are particularly active:
- The liver is the predominant site, responsible for approximately 50% of the body's synthesis.
- Other significant contributors include the intestine, the adrenal cortex, and the testes and ovaries.
Cellular Location of Enzymes:
The enzymatic machinery is distributed between two key cellular compartments:
- Enzymes for the initial stages are found in the cytoplasm.
- Enzymes for later stages are located within the membranes of the endoplasmic reticulum.
Requirements for Cholesterol Biosynthesis:
The synthesis of cholesterol is an energetically demanding process:
- Carbon Atoms: All 27 carbon atoms are derived from Acetyl-CoA. A total of 18 molecules are consumed.
- Reducing Equivalents: The process requires significant reducing power, supplied by NADPH (approx. 16 moles).
- Energy: The process requires considerable energy from ATP (approx. 36 moles).
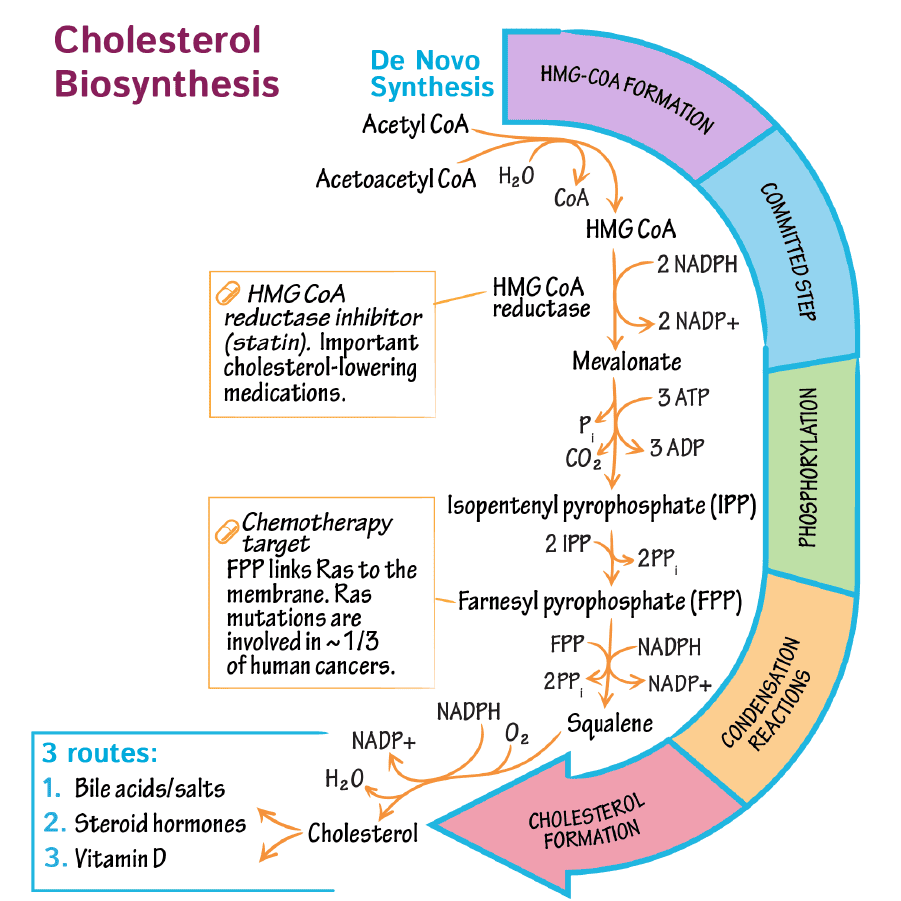
Steps of Cholesterol Biosynthesis: A Detailed Pathway
The complex pathway can be delineated into five principal stages:
Step 1: Formation of HMG-CoA
The synthesis initiates with the condensation of Acetyl-CoA units:
- Two molecules of Acetyl-CoA combine to form Acetoacetyl-CoA (catalyzed by thiolase).
- Acetoacetyl-CoA condenses with a third molecule of Acetyl-CoA to yield β-hydroxy-β-methylglutaryl-CoA (HMG-CoA), catalyzed by HMG-CoA synthase.
It is crucial to note the distinction from ketone body synthesis: the cytosolic HMG-CoA synthase produces HMG-CoA for cholesterol synthesis, while the mitochondrial HMG-CoA synthase participates in ketogenesis. This segregation ensures the pathways operate independently.
Step 2: Conversion of HMG-CoA to Mevalonate
This stage represents the rate-limiting and committed step in cholesterol biosynthesis:
- HMG-CoA is reduced to mevalonate in a reaction catalyzed by HMG-CoA reductase.
- This endoplasmic reticulum-bound enzyme requires two molecules of NADPH.
- HMG-CoA reductase is the primary therapeutic target for statins, a class of drugs that lower plasma cholesterol.
Step 3: Production of Activated Isoprenoid Units
Mevalonate is subsequently processed to generate activated 5-carbon units:
- Mevalonate undergoes a series of three phosphorylation steps, utilizing ATP.
- This is followed by decarboxylation to produce isopentenyl pyrophosphate (IPP), a 5-carbon isoprenoid unit.
- IPP can be isomerized to its structural cousin, dimethylallyl pyrophosphate (DMAPP).
Step 4: Synthesis of Squalene
The activated 5-carbon isoprenoid units are progressively linked:
- DMAPP condenses with IPP to form the 10-carbon geranyl pyrophosphate (GPP).
- GPP condenses with another IPP to yield the 15-carbon farnesyl pyrophosphate (FPP).
- Finally, two molecules of FPP condense head-to-head, mediated by squalene synthase and requiring NADPH, to form the 30-carbon linear molecule squalene.
Step 5: Conversion of Squalene to Cholesterol
The linear squalene molecule undergoes cyclization and a series of modifications:
- Squalene is first oxidized to squalene epoxide by squalene monooxygenase.
- Squalene epoxide undergoes a complex cyclization reaction to form lanosterol, the first true steroid compound in the pathway.
- From lanosterol, a multi-step process involving ~19 enzymatic reactions ensues, involving:
- Demethylation: Removal of three methyl groups, reducing the carbon count from 30 to 27.
- Double Bond Rearrangement: Shifts in the positions of double bonds.
- Reduction: Reduction of a double bond in the side chain.
These precise modifications culminate in the formation of cholesterol.
Regulation of Cholesterol Synthesis
The synthesis of cholesterol is a highly regulated process. The primary point of control is the enzyme HMG-CoA reductase, the rate-limiting step in the pathway. Regulation occurs through several sophisticated mechanisms:
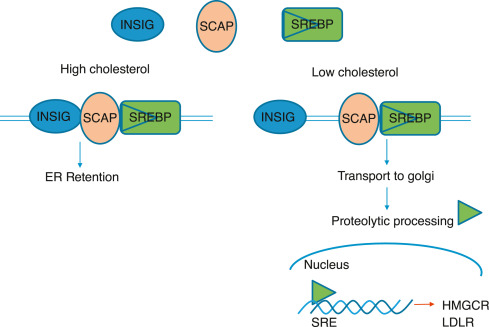
Transcriptional Control (Feedback Inhibition via SREBP Pathway):
- This is the most critical long-term regulatory mechanism.
- When intracellular cholesterol levels are high, they reduce the activity of SREBP-2 (Sterol Regulatory Element-Binding Protein 2).
- This leads to a reduction in the transcription of genes encoding HMG-CoA reductase and the LDL receptor, thus diminishing cholesterol synthesis.
- Conversely, low cellular cholesterol levels activate SREBP-2, promoting gene transcription and increasing both cholesterol synthesis and uptake.
Translational Control:
High concentrations of cholesterol also exert an inhibitory effect on the translation of HMG-CoA reductase mRNA, directly reducing the quantity of enzyme synthesized.
Enzyme Degradation (Proteolysis):
High sterol levels induce a conformational change in the reductase enzyme, making it more susceptible to ubiquitination and subsequent degradation by the proteasome. This shortens the enzyme's lifespan, leading to a quick reduction in its activity.
Covalent Modification (Hormonal Regulation and Energy Status):
HMG-CoA reductase exists in two interconvertible forms:
- Dephosphorylated form: More active.
- Phosphorylated form: Less active (inactive).
Phosphorylation is primarily mediated by AMP-activated protein kinase (AMPK), which is activated when cellular ATP is low. By phosphorylating and inactivating HMG-CoA reductase, AMPK conserves cellular energy.
Hormonal Influence:
- Insulin and Thyroid Hormones: Increase HMG-CoA reductase activity by promoting dephosphorylation (activation).
- Glucagon and Glucocorticoids: Decrease HMG-CoA reductase activity by promoting phosphorylation (inactivation).
Direct Inhibition by Drugs (Statins):
- Drugs such as lovastatin, simvastatin, and atorvastatin are competitive inhibitors of HMG-CoA reductase.
- They structurally resemble HMG-CoA and bind to the active site, blocking its ability to convert HMG-CoA to mevalonate and directly reducing the rate of cholesterol synthesis.
Inhibition by Bile Acids:
Bile acids, which are derivatives of cholesterol, can also contribute to feedback regulation by inhibiting HMG-CoA reductase activity.
Connecting to Cholesterol Transport:
While not a direct regulatory mechanism for synthesis, the major players in cholesterol transport are intrinsically linked to overall cholesterol homeostasis:
- Low-Density Lipoproteins (LDL): Primarily transport cholesterol from the liver to peripheral tissues. Often called "bad cholesterol."
- High-Density Lipoproteins (HDL): Transport excess cholesterol from peripheral tissues back to the liver for excretion or recycling (reverse cholesterol transport). Often called "good cholesterol."
Formation and Excretion of Bile Acids and Bile Salts
Bile acids are a family of steroid acids that represent the major catabolic products of cholesterol in the body. Their primary physiological function is to facilitate the digestion and absorption of dietary fats and fat-soluble vitamins in the small intestine. They also play a crucial role in cholesterol homeostasis by being the principal route for cholesterol excretion.
What is Bile?
Bile is a complex, watery, yellowish-green fluid produced by the liver. It consists of a watery mixture of organic and inorganic compounds.
The quantitatively most important organic components of bile are phosphatidylcholine (lecithin) and conjugated bile salts.
Bile can either pass directly from the liver into the duodenum (the first part of the small intestine) via the common bile duct, or it can be stored and concentrated in the gallbladder when not immediately needed for digestion.
A. Synthesis of Primary Bile Acids
The synthesis of bile acids, known as cholic acid and chenodeoxycholic acid, occurs exclusively in the liver. This multi-step pathway converts the hydrophobic cholesterol molecule into more polar, amphipathic bile acids, making them water-soluble.
Initiation - The Rate-Limiting Step:
The synthesis pathway involves the insertion of hydroxyl groups at specific positions on the steroid structure of cholesterol. The hydrocarbon chain is also shortened by three carbons.
The first and rate-limiting step in bile acid synthesis is the introduction of a hydroxyl group at carbon 7 of cholesterol, forming 7α-hydroxycholesterol.
This reaction is catalyzed by the enzyme cholesterol 7α-hydroxylase (CYP7A1).
CYP7A1 is a cytochrome P450 enzyme, requiring molecular oxygen (O₂) and NADPH.
Regulation: The activity of CYP7A1 is highly regulated. It is inhibited by bile acids (a feedback mechanism) and induced by cholesterol (when cholesterol levels are high). This ensures that bile acid synthesis is responsive to both bile acid demand and cholesterol availability.
Subsequent Reactions:
Following the initial hydroxylation, 7α-hydroxycholesterol undergoes a series of additional modifications. These steps involve:
- Further hydroxylations (e.g., at C-12 to form cholic acid, which is a triol - having three hydroxyl groups).
- Epimerization of the 3β-hydroxyl group to a 3α-hydroxyl group.
- Reduction of the double bond in the B ring.
- Oxidation of the side chain (carbon atoms 24, 25, 26, and 27) and its cleavage to introduce a carboxyl group at C-24, shortening the side chain from 8 to 5 carbons.
Formation of Primary Bile Acids:
These reactions ultimately lead to the formation of the two primary bile acids:
- Cholic acid: (a triol) Has hydroxyl groups at C-3α, C-7α, and C-12α.
- Chenodeoxycholic acid: (a diol) Has hydroxyl groups at C-3α and C-7α.
B. Conjugation of Primary Bile Acids to Form Bile Salts
To significantly improve their ability to emulsify fat and enhance their water solubility, primary bile acids are further modified in the liver through a process called conjugation. They are joined with either the amino acid glycine or taurine.
Mechanism:
The carboxyl group (–COOH) at the end of the bile acid side chain forms an amide bond with the amino group (–NH₂) of glycine or taurine.
This reaction is catalyzed by bile acid-CoA ligase (which activates the bile acid by forming a CoA thioester) and bile acid-CoA:amino acid N-acyltransferase.
Resulting Conjugated Bile Acids (Bile Salts):
This generates the conjugated bile acids:
- Taurocholic acid and Taurochenodeoxycholic acid
- Glycocholic acid and Glycocholic acid
These conjugated forms are all necessary to give bile its essential function in fat digestion.
At physiological pH, these conjugated bile acids exist as anions (negatively charged) due to the low pKa of their conjugates. Therefore, they are referred to as bile salts (e.g., taurocholate, glycocholate). The term "bile salts" specifically refers to these ionized forms.
Physiological Significance of Conjugation:
- Increased Solubility & Emulsification: Conjugation makes bile acids much more soluble and improves their amphipathic nature, crucial for emulsifying dietary fats.
- Effective Detergency: The salts are large, negatively charged ions that are not readily absorbed by passive diffusion in the upper region of the small intestine, ensuring sustained activity.
- PKA Reduction: Conjugation lowers the pKa of the bile acids, ensuring that they remain ionized (charged) even in the acidic environment of the upper small intestine.
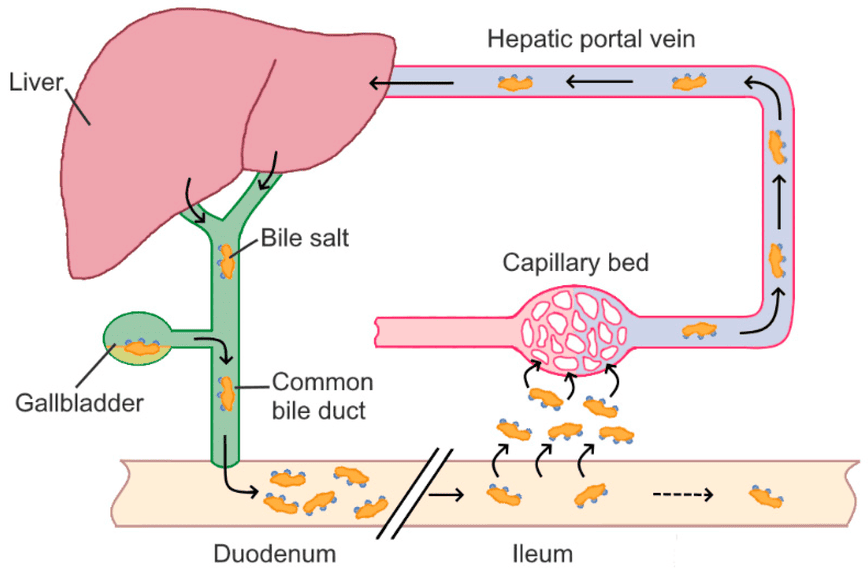
C. Enterohepatic Circulation of Bile Salts
Bile salts are essential for fat digestion, but the body has a highly efficient system to conserve them rather than synthesizing new ones for every meal. This system is called the enterohepatic circulation.
Secretion:
Synthesized and conjugated bile salts are secreted from the liver, stored in the gallbladder, and released into the duodenum after a fatty meal.
Function in Small Intestine:
In the duodenum and jejunum, bile salts emulsify dietary fats and form mixed micelles.
Reabsorption:
A remarkable 95% of bile salts are reabsorbed in the ileum (the final part of the small intestine). This reabsorption occurs via a specialized, active transport system known as the apical sodium-dependent bile acid transporter (ASBT) in the ileal enterocytes. Some passive reabsorption of unconjugated bile acids can also occur in the jejunum and colon.
Portal Vein Transport:
Once reabsorbed, bile salts enter the portal venous blood and are transported back to the liver, mostly bound to albumin.
Hepatic Uptake:
The liver efficiently extracts the bile salts from the portal blood via specific transporters.
Recycling:
The liver then re-secretes these reabsorbed bile salts into the bile, completing the circulation. This cycle can occur 4-12 times a day.
D. Formation and Excretion of Secondary Bile Acids
Not all bile acids are reabsorbed directly. Bacterial action in the gut leads to the formation of secondary bile acids.
Bacterial Deconjugation:
As bile salts travel through the colon, intestinal bacteria can deconjugate them, removing glycine or taurine.
Bacterial Dehydroxylation:
These free primary bile acids can then be further metabolized by gut bacteria, specifically undergoing 7α-dehydroxylation. This results in the formation of secondary bile acids:
- Deoxycholic acid (from cholic acid)
- Lithocholic acid (from chenodeoxycholic acid)
Fate of Secondary Bile Acids:
Most secondary bile acids are also reabsorbed and return to the liver. In the liver, deoxycholic acid can be re-conjugated. Lithocholic acid, which is less soluble, is often sulfonated before being secreted back into bile, which aids in its excretion.
E. Excretion of Cholesterol
The excretion of cholesterol from the body primarily occurs via two main routes:
- Conversion to Bile Acids and Excretion: A small fraction of bile salts (about 5%, or 0.2-0.6 grams per day) is not reabsorbed and is instead excreted in the feces. This represents a net loss and is the most significant route for cholesterol elimination.
- Direct Secretion of Unesterified Cholesterol into Bile: The liver can also secrete free, unesterified cholesterol directly into the bile. A portion of this is reabsorbed, but a significant amount is excreted. If the concentration of cholesterol in bile exceeds the solubilizing capacity of bile salts, it can precipitate, leading to cholesterol gallstones.
Synthesis of Steroid Hormones
Cholesterol is not merely a structural component of cell membranes or a precursor for bile acids; it is also the obligate precursor for all steroid hormones. These powerful signaling molecules regulate a vast array of physiological processes, including metabolism, inflammation, immune responses, salt and water balance, sexual development, and reproduction.
A. General Pathway for Steroid Hormone Synthesis
The synthesis of all steroid hormones follows a common, fundamental pathway that begins with cholesterol. This process primarily occurs in the mitochondria and endoplasmic reticulum of steroidogenic tissues.
Tissue-Specific Synthesis:
While virtually all cells contain cholesterol, steroid hormone synthesis is restricted to specialized endocrine tissues, including:
- Adrenal cortex: Produces glucocorticoids, mineralocorticoids, and some adrenal androgens.
- Gonads (Testes and Ovaries): Produce androgens, estrogens, and progestins.
- Placenta: Produces estrogens and progestins during pregnancy.
Rate-Limiting Step: Transport into Mitochondria:
- The first and rate-limiting step is the transport of cholesterol from the cytoplasm into the inner mitochondrial membrane.
- This transport is mediated by the Steroidogenic Acute Regulatory protein (StAR). StAR's activity is rapidly regulated by tropic hormones (e.g., ACTH, LH).
Initial Conversion: Cholesterol to Pregnenolone:
- Once inside the inner mitochondrial membrane, cholesterol is converted to pregnenolone.
- This is catalyzed by the cholesterol desmolase complex, also known as P450scc (cytochrome P450 side-chain cleavage enzyme), which requires NADPH and O₂.
- Pregnenolone is the universal precursor for all other steroid hormones.
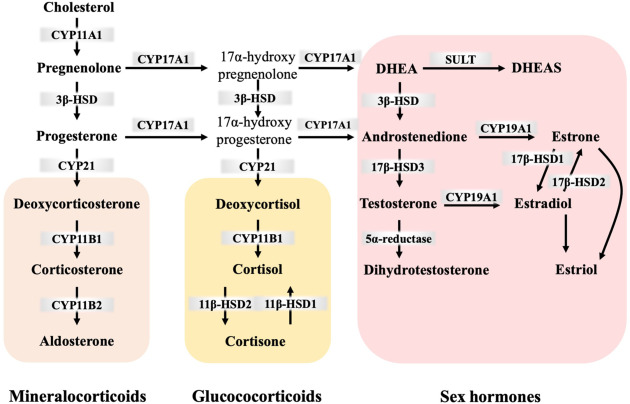
B. Major Classes of Steroid Hormones
From pregnenolone, the pathway diverges. The specific hormones produced depend on the enzymatic machinery present in the particular tissue.
Progestins (C21 Steroids):
- Progesterone is derived directly from pregnenolone.
- Function: Crucial for maintaining pregnancy and regulating the menstrual cycle.
- Primary site of synthesis: Ovaries (corpus luteum), adrenal cortex, placenta.
Glucocorticoids (C21 Steroids):
- Cortisol is the primary human glucocorticoid.
- Pathway: Pregnenolone → Progesterone → ... → Cortisol.
- Function: Regulates metabolism, suppresses immune responses, reduces inflammation, and helps adapt to stress.
- Primary site of synthesis: Adrenal cortex (zona fasciculata).
Mineralocorticoids (C21 Steroids):
- Aldosterone is the most potent human mineralocorticoid.
- Pathway: Pregnenolone → Progesterone → ... → Aldosterone.
- Function: Regulates electrolyte balance by promoting sodium reabsorption and potassium excretion, thus influencing blood pressure.
- Primary site of synthesis: Adrenal cortex (zona glomerulosa).
Androgens (C19 Steroids):
- Dehydroepiandrosterone (DHEA), Androstenedione, and Testosterone are key androgens.
- Function: Responsible for the development of male secondary sexual characteristics and libido in both sexes.
- Primary site of synthesis: Testes, adrenal cortex, ovaries.
Estrogens (C18 Steroids):
- Estradiol is the most potent and abundant human estrogen.
- Pathway: Estrogens are synthesized from androgens (testosterone or androstenedione) through a reaction catalyzed by the enzyme aromatase.
- Function: Responsible for the development of female secondary sexual characteristics and regulation of the menstrual cycle.
- Primary site of synthesis: Ovaries, placenta, adipose tissue, testes (to a lesser extent).
C. Regulation of Steroid Hormone Synthesis
The synthesis is tightly regulated by the hypothalamic-pituitary-adrenal/gonadal axes.
- Tropic Hormones: Specific peptide hormones from the anterior pituitary stimulate target endocrine glands:
- Adrenocorticotropic hormone (ACTH): Stimulates the adrenal cortex (cortisol).
- Luteinizing hormone (LH): Stimulates testosterone production in testes and progesterone in ovaries.
- Follicle-stimulating hormone (FSH): Stimulates estrogen production by ovarian follicles.
- Feedback Inhibition: High levels of circulating steroid hormones typically exert negative feedback on the hypothalamus and pituitary gland.
- Enzyme Specificity: The expression and activity of specific steroidogenic enzymes (e.g., 21-hydroxylase, aromatase) within different tissues dictate which hormones are produced. Genetic deficiencies can lead to disorders like congenital adrenal hyperplasia.
Transport of Cholesterol by Lipoproteins
Cholesterol, being a lipid, is largely insoluble in the aqueous environment of blood plasma. To be efficiently transported between tissues for synthesis, utilization, and excretion, cholesterol (along with other lipids like triglycerides and phospholipids) is packaged into complex particles called lipoproteins. These molecular transporters have a hydrophilic exterior and a hydrophobic core, allowing them to carry lipids through the bloodstream.
Cholesteryl ester in the diet is hydrolyzed to cholesterol, which is then absorbed by the intestine together with dietary unesterified cholesterol and other lipids. It is then incorporated into chylomicrons.
Ninety-five percent of the chylomicron cholesterol is delivered to the liver in chylomicron remnants. Most of the cholesterol secreted by the liver in VLDL is retained during the formation of IDL and ultimately LDL, which is taken up by the LDL receptor in liver and extrahepatic tissues.
A. Structure of Lipoproteins
All lipoproteins share a common structural organization:
- Hydrophobic Core: Contains the most water-insoluble lipids:
- Triglycerides (TGs)
- Cholesteryl esters (CEs)
- Hydrophilic Shell: Surrounds the core and allows the particle to be soluble in blood:
- Phospholipids
- Free (unesterified) Cholesterol
- Apolipoproteins: Proteins integral to the shell that provide structural integrity, act as enzyme cofactors (e.g., ApoC-II), and serve as ligands for cell surface receptors (e.g., ApoB-100).
B. Classes of Lipoproteins
Lipoproteins are classified based on their density (more lipid = less dense). From largest/least dense to smallest/most dense, the main classes are:
Chylomicrons (CM):
- Origin: Intestine.
- Primary Lipid: Dietary triglycerides (>80%).
- Main Apolipoproteins: ApoB-48, ApoC-II, ApoE.
- Function: Transport dietary (exogenous) lipids from the intestine to peripheral tissues and then to the liver.
- Metabolism: Nascent chylomicrons acquire ApoC-II and ApoE from HDL. ApoC-II activates lipoprotein lipase (LPL) in capillaries, which hydrolyzes TGs. The resulting chylomicron remnants, enriched in cholesteryl esters and ApoE, are taken up by the liver.
Very Low-Density Lipoproteins (VLDL):
- Origin: Liver.
- Primary Lipid: Endogenously synthesized triglycerides (50-60%).
- Main Apolipoproteins: ApoB-100, ApoC-II, ApoE.
- Function: Transport endogenously synthesized lipids from the liver to peripheral tissues.
- Metabolism: Similar to chylomicrons, LPL hydrolyzes TGs from VLDL. As VLDL loses TGs, it becomes smaller and denser, first forming intermediate-density lipoproteins (IDL).
Intermediate-Density Lipoproteins (IDL):
- Origin: Formed from VLDL after triglyceride hydrolysis.
- Primary Lipid: Roughly equal amounts of TGs and cholesteryl esters.
- Main Apolipoproteins: ApoB-100, ApoE.
- Function: An intermediate in the conversion of VLDL to LDL. About half are taken up by the liver, and the rest are metabolized to LDL.
Low-Density Lipoproteins (LDL):
- Origin: Primarily from the catabolism of VLDL and IDL.
- Primary Lipid: Cholesteryl esters (~45-50%).
- Main Apolipoprotein: ApoB-100.
- Function: Transport cholesterol from the liver to peripheral tissues. Often called "bad cholesterol."
- Metabolism: Cells needing cholesterol take up LDL particles via the LDL receptor, which specifically recognizes ApoB-100.
High-Density Lipoproteins (HDL):
- Origin: Liver and intestine.
- Primary Lipid: Relatively rich in protein (~50%) and phospholipids.
- Main Apolipoproteins: ApoA-I (major), ApoC-II, ApoE.
- Function: Often called "good cholesterol." HDL plays a crucial role in reverse cholesterol transport, collecting excess cholesterol from peripheral cells and transporting it back to the liver. It also serves as a reservoir for ApoC-II and ApoE.
- Metabolism: Nascent HDL particles acquire free cholesterol from peripheral cells. This cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT). Mature HDL can exchange cholesteryl esters for TGs with other lipoproteins (via CETP) and is finally taken up by the liver (via SR-B1).
C. Exogenous vs. Endogenous Pathways of Lipid Transport
- Exogenous Pathway: Deals with dietary lipids. Chylomicrons are the key lipoproteins.
- Endogenous Pathway: Deals with endogenously synthesized lipids. VLDL, IDL, and LDL are the main players for distribution, while HDL mediates reverse transport.
D. Role of Key Enzymes and Receptors
- Lipoprotein Lipase (LPL): Hydrolyzes TGs in chylomicrons and VLDL in capillaries.
- Hepatic Lipase (HL): Hydrolyzes TGs and phospholipids in IDL and HDL.
- Lecithin-Cholesterol Acyltransferase (LCAT): Esterifies free cholesterol to cholesteryl esters within HDL.
- Cholesteryl Ester Transfer Protein (CETP): Facilitates the exchange of cholesteryl esters and TGs between lipoproteins.
- LDL Receptor: Mediates the uptake of LDL (and IDL) via recognition of ApoB-100.
- Scavenger Receptor Class B Type 1 (SR-B1): Mediates selective uptake of cholesteryl esters from HDL by the liver.
Regulation of Cholesterol Synthesis, Metabolism, and Transport
Maintaining cholesterol homeostasis is critical. The body employs an intricate network of regulatory mechanisms, with the primary point of control being the enzyme HMG-CoA reductase.
A. Regulation of Cholesterol Synthesis
Transcriptional Control (Gene Expression):
- SREBP-2 (Sterol Regulatory Element-Binding Protein-2): This is the master regulator.
- Low Cellular Cholesterol: SREBP-2 is cleaved, moves to the nucleus, and binds to Sterol Regulatory Elements (SREs). This activates transcription of genes for HMG-CoA reductase and the LDL receptor, increasing both cholesterol production and uptake.
- High Cellular Cholesterol: SREBP-2 is retained in the ER, and transcription is suppressed.
Post-Translational Control (Enzyme Activity & Degradation):
- Phosphorylation (Covalent Modification):
- AMP-activated protein kinase (AMPK): When cellular energy is low, AMPK phosphorylates and inactivates HMG-CoA reductase.
- Insulin: Promotes dephosphorylation, activating the enzyme.
- Glucagon: Promotes phosphorylation, inactivating the enzyme.
- Proteasomal Degradation: High levels of cholesterol cause HMG-CoA reductase to be degraded by the proteasome.
B. Regulation of Bile Acid Synthesis
- Cholesterol 7α-hydroxylase (CYP7A1): This is the rate-limiting enzyme.
- Negative Feedback by Bile Acids: High levels of bile acids returning to the liver inhibit the transcription of the CYP7A1 gene.
- Positive Regulation by Cholesterol: Increased cholesterol induces CYP7A1 activity, ensuring excess cholesterol can be eliminated.
C. Regulation of Cholesterol Transport
LDL Receptor Regulation:
- The number of LDL receptors on the cell surface is the primary determinant of LDL clearance from the blood.
- High Cellular Cholesterol: Downregulates LDL receptor synthesis (via the SREBP-2 mechanism).
- Low Cellular Cholesterol: Upregulates LDL receptor synthesis.
- PCSK9: This enzyme promotes the degradation of the LDL receptor. Inhibitors of PCSK9 are a new class of drugs for lowering LDL.
Dietary and Hormonal Factors:
- Saturated and Trans Fats: Increase LDL cholesterol.
- Soluble Fiber: Lowers LDL cholesterol by interfering with bile acid reabsorption.
- Insulin: Promotes cholesterol synthesis.
- Estrogens: Tend to lower LDL and raise HDL.
D. Importance of Cholesterol Homeostasis
The tight regulation is vital because both insufficient (hypocholesterolemia) and excessive (hypercholesterolemia) cholesterol levels are detrimental. Excess cholesterol, particularly carried by LDL, can lead to its deposition in arterial walls, causing atherosclerosis.
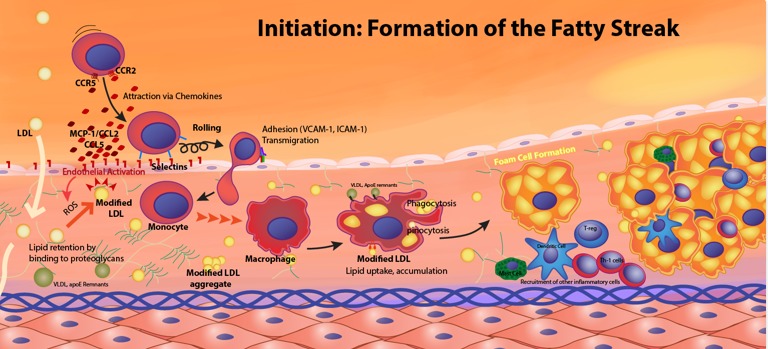
Clinical Significance of Cholesterol: Atherosclerosis
Atherosclerosis is a chronic inflammatory disease characterized by the buildup of fatty plaques within the arterial walls, leading to hardening and narrowing of the arteries.
A. Hypercholesterolemia and Dyslipidemia
- Hypercholesterolemia: Abnormally high levels of cholesterol in the blood.
- Dyslipidemia: A broader term for abnormal lipid levels, including high LDL ("bad cholesterol"), low HDL ("good cholesterol"), and high triglycerides.
B. The Role of Lipoproteins in Atherosclerosis
- Low-Density Lipoprotein (LDL) - The Primary Atherogenic Particle:
- High LDL levels lead to its infiltration into the arterial wall, where it becomes oxidized (oxLDL).
- Macrophages ingest oxLDL in an uncontrolled manner, transforming into foam cells.
- Accumulations of foam cells form fatty streaks, the earliest lesions of atherosclerosis.
- High-Density Lipoprotein (HDL) - The Anti-Atherogenic Particle:
- HDL is crucial for reverse cholesterol transport, removing excess cholesterol from arterial walls and transporting it to the liver.
- HDL also has antioxidant and anti-inflammatory properties. High HDL levels are associated with reduced CVD risk.
C. Pathogenesis of Atherosclerosis
The development of atherosclerotic plaques is a multi-stage process:
- Endothelial Dysfunction: Damage to the artery's inner lining.
- LDL Infiltration and Oxidation: LDL enters the arterial wall and becomes oxidized.
- Macrophage Recruitment and Foam Cell Formation: Immune cells are recruited and become lipid-laden foam cells.
- Smooth Muscle Cell Migration and Proliferation: These cells contribute to the bulk of the plaque.
- Fibrous Cap Formation: A cap of collagen and smooth muscle cells forms over the lipid core.
- Plaque Progression and Complications: Over time, plaques can grow, develop a necrotic core, and become unstable. Plaque rupture exposes the core to blood, leading to rapid thrombus (blood clot) formation, which can cause a heart attack or stroke.
D. Management of Dyslipidemia and CVD Risk
- Lifestyle Modifications: Diet, physical activity, weight management, and smoking cessation.
- Pharmacological Interventions:
- Statins (HMG-CoA Reductase Inhibitors): The most effective drugs for lowering LDL.
- Ezetimibe: Inhibits cholesterol absorption.
- PCSK9 Inhibitors: Prevent LDL receptor degradation.
- Bile Acid Sequestrants: Increase cholesterol excretion via bile acids.
Biochemistry: Cholesterol Metabolism
Test your knowledge with these 40 questions.
Cholesterol Metabolism Quiz
Question 1/40
Quiz Complete!
Here are your results, .
Your Score
38/40
95%