Analysis of Clinical Case: Sickle Cell Disease
Clinical Scenario
A 2-year-old boy from Mukono district presents with recurrent episodes of severe bone pain (hands, feet, and sternum pain), jaundice, and fatigue for 3 days.
Laboratory findings reveal:
- Haemoglobin = 6.2 g/dL (normal range: 11-16 g/dL)
- Peripheral smear: sickled red blood cells
- Liver function tests: Elevated bilirubin
- Haemoglobin electrophoresis test of his blood shows increased percentage of sickled haemoglobin (HbS)
A diagnosis of Vaso-occlusive crisis, and severe anaemia in Sickle Cell Disease was made.
- Clinical Signs: Recurrent severe bone pain (vaso-occlusive crisis), jaundice (evidence of hemolysis), and fatigue (symptom of anaemia).
- Laboratory Findings: Low haemoglobin (severe anaemia), sickled red blood cells on peripheral smear, elevated bilirubin (confirming high rate of cell breakdown), and definitive identification of sickled haemoglobin (HbS) via electrophoresis.
(a) The Amino Acid Change in Haemoglobin (HbS)
This part requires a detailed breakdown of the specific molecular error in the patient's haemoglobin protein, focusing on the identity of the amino acids and the genetic origin of the mistake.
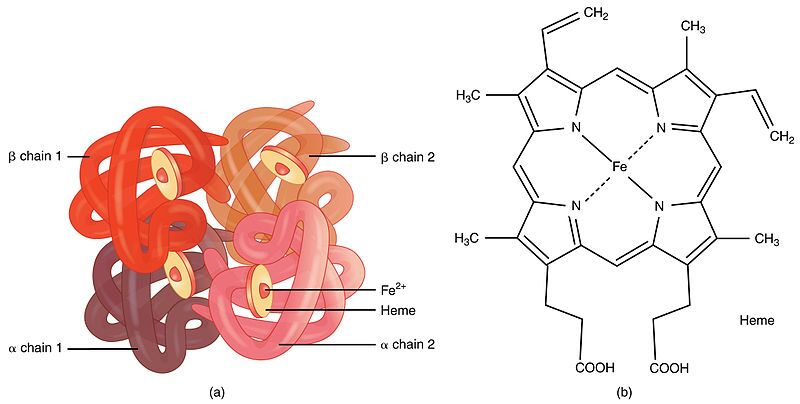
Step 1: Introduction to Haemoglobin Structure
First, it's important to understand what haemoglobin is. Haemoglobin is the primary protein found within red blood cells (erythrocytes) and its main function is to transport oxygen from the lungs to the body's tissues. It is a large, complex protein with a quaternary structure, meaning it is composed of multiple polypeptide subunits. A normal adult haemoglobin molecule (HbA) is a tetramer, consisting of four chains: two identical alpha (α)-globin chains and two identical beta (β)-globin chains. The genetic defect in sickle cell disease specifically affects the gene that provides the instructions for the beta-globin chain.

Step 2: The Specific Amino Acid Substitution
The defining molecular event in sickle cell disease is a single amino acid substitution at a precise location within the beta-globin polypeptide chain.
In a person with normal adult haemoglobin (HbA), the amino acid at the sixth position from the beginning (the N-terminus) of the beta-globin chain is Glutamic Acid (abbreviated as Glu or E).
In this patient with sickle cell disease, the haemoglobin is abnormal (called HbS). At that exact same sixth position, the Glutamic Acid has been replaced by the amino acid Valine (abbreviated as Val or V).
This single change, Glu6Val, is the sole cause of the disease.

Step 3: The Chemical Nature of the Amino Acids Involved
The severity of this substitution is due to the drastically different chemical "personalities" of the R-groups (side chains) of Glutamic Acid and Valine. This position is on the outer surface of the protein, where it is exposed to the watery environment inside the red blood cell.
| Amino Acid | Chemical Class & Properties | Behavior in Water |
|---|---|---|
| Glutamic Acid (Normal) | Its side chain contains a carboxyl group (`-CH₂-CH₂-COOH`). At the neutral pH inside a red blood cell (~7.4), this group loses a proton and becomes negatively charged (`-COO⁻`). Therefore, it is an acidic, polar, and charged amino acid. | Because it is charged and polar, Glutamic Acid is hydrophilic ("water-loving"). It forms favorable interactions with polar water molecules and is perfectly stable on the protein's surface. |
| Valine (Mutant) | Its side chain is an isopropyl group (`-CH(CH₃)₂`), which is a small, branched structure made only of carbon and hydrogen. These bonds are nonpolar. Therefore, Valine is a nonpolar, aliphatic, and neutral amino acid. | Because it is nonpolar, Valine is hydrophobic ("water-fearing"). It is thermodynamically unfavorable for this "oily" side chain to be exposed to water. It will seek to interact with other nonpolar groups to hide from the aqueous environment. |
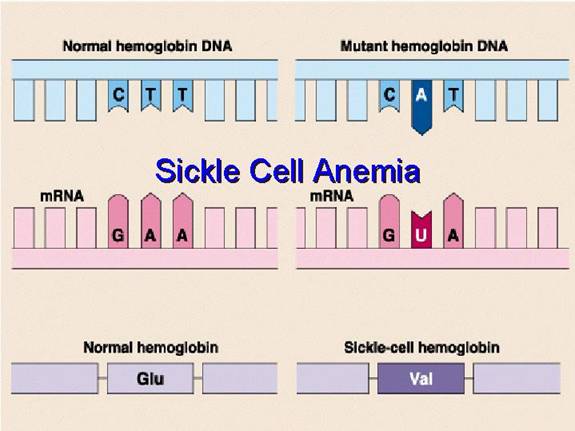
Step 4: The Chemical Basis of the Mutation (Genetics)
This amino acid error originates from a single change in the DNA sequence of the beta-globin gene. This type of mutation is called a point mutation, specifically a missense mutation because it results in a codon that codes for a different amino acid.
- The DNA Code: The genetic code is read in triplets called codons. The DNA codon on the template strand that codes for Glutamic Acid at position 6 is CTC. The corresponding codon on the coding strand is GAG.
- The Mutation: A single nucleotide change occurs where the Adenine (A) in the middle of the GAG codon is substituted for a Thymine (T). This is known as a transversion (a purine is replaced by a pyrimidine).
- Transcription to mRNA: The mutated DNA codon, now GTG on the coding strand, is transcribed into a messenger RNA (mRNA) codon. The mRNA codon becomes GUG.
- Translation to Protein: During protein synthesis at the ribosome, the cellular machinery reads the GUG codon and inserts the amino acid Valine into the growing polypeptide chain instead of Glutamic Acid.
Therefore, a single DNA base change leads to a single mRNA codon change, which in turn leads to the single, catastrophic amino acid substitution that defines sickle cell disease.
(b) Pathophysiology: From Molecular Defect to Clinical Symptoms
This section explains the step-by-step process of how the single Glu6Val substitution causes the haemoglobin to malfunction and leads to the patient's observed symptoms.

Step 1: The Molecular Effect - Polymerization of Deoxy-HbS
The key event is the behavior of HbS when it is in the deoxygenated state. In the oxygenated state (in the lungs), HbS functions almost normally as an oxygen carrier.
- Conformational Change: When a red blood cell travels to peripheral tissues and releases oxygen, the haemoglobin tetramer shifts from a high-oxygen-affinity "R-state" (relaxed) to a low-oxygen-affinity "T-state" (tense).
- Exposure of the Hydrophobic Patch: In HbS, this shift to the T-state causes a structural change that exposes the hydrophobic Valine at position β6 on the protein's surface. This creates a "sticky patch."
- Intermolecular Interaction: This exposed, oily Valine seeks to escape the aqueous cytosol. Coincidentally, the T-state conformation of another HbS molecule creates a complementary hydrophobic pocket on its surface. The Valine from one HbS molecule fits perfectly into this pocket on another HbS molecule.
- Polymerization: This initial binding is the critical step that seeds the formation of long, rigid polymers. HbS molecules begin to aggregate in a highly ordered fashion, forming long, insoluble fibers that can contain millions of haemoglobin molecules.
Step 2: The Cellular Effect - Erythrocyte Sickling
Shape Distortion: These long, stiff haemoglobin polymers grow to be longer than the diameter of the red blood cell itself. They physically push against the cell membrane from the inside, distorting the cell from its normal, flexible biconcave disc shape into a rigid, elongated, crescent or "sickle" shape.
Loss of Deformability: This sickling process causes a dramatic loss of the cell's flexibility. It becomes hard and unable to deform. This process is initially reversible if the cell becomes reoxygenated, but repeated sickling events cause permanent membrane damage, leading to irreversibly sickled cells.
Step 3: Connecting to the Clinical Manifestations
The physical properties of these sickled cells are directly responsible for the patient's symptoms:
- Vaso-occlusive Crisis (Severe Bone Pain): The rigidity and abnormal shape of the sickled cells prevent them from navigating the narrow microvasculature (capillaries). They get stuck, leading to vascular occlusion. This "logjam" blocks blood flow, causing severe tissue ischemia (lack of oxygen). The resulting hypoxia and infarction trigger intense inflammatory responses and severe pain. This is the cause of the boy's pain in his hands, feet, and sternum, which are common sites for such crises.
- Severe Anaemia (Fatigue): The sickled cells are mechanically fragile. The membrane is damaged by the internal polymers and by the stress of passing through the circulation. These cells are recognized by the reticuloendothelial system (macrophages in the spleen and liver) and are destroyed prematurely. This process, called extravascular hemolysis, reduces the average red blood cell lifespan from a normal 120 days to a mere 10-20 days. The bone marrow's production of new cells cannot keep up with this high rate of destruction, leading to a state of chronic hemolytic anaemia. The patient's very low haemoglobin level of 6.2 g/dL is a direct measure of this. The reduced oxygen-carrying capacity of the blood results in the profound fatigue.
- Jaundice (Elevated Bilirubin): The massive and continuous breakdown of red blood cells (hemolysis) leads to the release of large amounts of haemoglobin. The heme portion is catabolized into bilirubin. This high rate of bilirubin production overwhelms the liver's ability to conjugate it for excretion. The resulting buildup of unconjugated bilirubin in the bloodstream leads to hyperbilirubinemia, which manifests clinically as jaundice (yellowing of the skin and sclera), confirmed by the lab results.
(c) Therapeutic Approaches Based on Amino Acid Chemistry
Knowing that the core problem is a hydrophobic amino acid causing polymerization allows for the design of targeted therapies.
Strategy 1: Altering the Amino Acid Composition Inside the Cell
This approach aims to reduce the relative concentration of the problematic HbS.
- Induction of Fetal Haemoglobin (HbF): Fetal haemoglobin (HbF) is composed of α₂γ₂ chains. The gamma (γ)-globin chain does not have Valine at position 6 and does not participate in polymerization. Pharmacological agents like hydroxyurea can reactivate the expression of the γ-globin gene in adults. By increasing the amount of HbF inside the red blood cell, the concentration of HbS is effectively diluted. The presence of HbF molecules physically interferes with the aggregation of HbS molecules, acting as a potent polymerization inhibitor. This is a direct manipulation of the cell's overall haemoglobin amino acid profile to mitigate the effects of the faulty beta chain.
Strategy 2: Directly Targeting the Unfavorable Amino Acid Interaction
This is the most direct chemical approach, aiming to stop the Valine from interacting with its target.
- Polymerization Inhibitors: The goal is to design a molecule that prevents the key hydrophobic interaction. This can be done in several ways:
- Capping the Valine: A drug could be designed to bind directly to the exposed hydrophobic Valine at position β6, making it unavailable to interact with other molecules.
- Blocking the Pocket: A drug could bind to the complementary hydrophobic pocket on an adjacent HbS molecule, preventing the Valine from docking there.
- Altering the Conformation: A class of drugs called allosteric modulators, such as Voxelotor, binds to haemoglobin and increases its affinity for oxygen. This stabilizes the molecule in the oxygenated R-state, even at lower oxygen levels. Since polymerization only occurs in the deoxygenated T-state, this prevents the Valine from being exposed in the first place, thus inhibiting sickling. This is a therapy based entirely on manipulating the protein's shape, which is dictated by its amino acid chemistry.
Strategy 3: Correcting the Amino Acid Code at the Genetic Level
This is the most fundamental approach, aiming to fix the DNA instruction so the correct amino acid is made.
- Gene Therapy/Gene Editing: This therapeutic strategy bypasses the protein problem by going to the source. Using technologies like CRISPR-Cas9, it is possible to edit the patient's hematopoietic stem cells. The goal is to revert the mutated DNA codon GTG back to the normal GAG. By correcting the genetic blueprint, the cell's machinery will once again transcribe a GAG codon into the mRNA and translate it into Glutamic Acid. This restores the normal, hydrophilic amino acid to position 6, completely eliminating the chemical basis for polymerization and offering a potential cure for the disease.