Mechanism of Drug Action (Pharmacodynamics)
Pharmacodynamics is the study of how drugs interact with the body at a molecular level. By the end of this guide, you will master:
- The four primary protein targets for drugs: Receptors, Ion Channels, Enzymes, and Transporters.
- The specific properties of receptors, including affinity, intrinsic activity, potency, and efficacy.
- The exact definitions of agonists (full, partial, inverse) and antagonists.
- The four major families of receptors and their specific operating speeds and mechanisms.
- A detailed understanding of G-Protein-Coupled Receptors (GPCRs) and their internal signaling pathways (cAMP and IP3/DAG).
Introduction to Pharmacodynamics
Pharmacodynamics is the branch of pharmacology concerned exclusively with the actions, interactions, and the specific mechanism (or mode) of action of drugs within the body. In simple terms, it studies exactly what the drug does to the body to produce a biological effect.
When a drug enters the body, it must interact with something to cause a change. These interactions fall into two broad categories:
- Highly Specific Interactions: The drug precisely binds to a specific biological target (most commonly a pharmacological receptor) to exert its effect.
- Non-Specific Interactions: The drug produces an effect without binding to a specific receptor. For example, an antacid (like calcium carbonate) simply neutralizes stomach acid through basic chemistry, without needing a receptor.
Molecular & Biochemical Mechanisms of Drug Action
For drugs that act specifically, they must bind to certain proteins on or inside mammalian cells. These protein targets can be broadly divided into four fundamental categories:
- Receptors
- Ion Channels
- Enzymes
- Carrier Molecules (Transporters)
Let us examine each of these four targets in deep detail, including the exact drugs that target them.
Target I: Receptors
Receptors are highly specialized protein structures located either on the surface of the mammalian cell membrane or entirely within the cell.
They act as the sensing elements in the chemical communication system that coordinates the functions of all the different cells in the body. Natural chemical messengers (endogenous ligands) bind to these receptors to tell the cell what to do. These natural messengers include:
- Hormones (e.g., insulin, estrogen).
- Neurotransmitters (e.g., acetylcholine, dopamine).
- Other local mediators / Autocoids (e.g., Histamine, Serotonin / 5-HT).
Many therapeutically useful drugs work by hijacking this system. They act either as agonists (mimicking the natural messenger) or antagonists (blocking the natural messenger) on these known endogenous receptors.
Key Characteristics of Drugs Acting via Receptors
- Low Concentrations: Because receptors are highly sensitive, drugs targeting them can act effectively at very low concentrations in the blood.
- Structure–Activity Relationship (SAR): Receptors are extremely picky about shape. Very small modifications to a drug's functional chemical groups, stereochemistry (3D arrangement), or molecular shape can significantly impact how tightly the drug binds (binding affinity) and how well it works (pharmacological activity).
- Specific Antagonism: Their effects can be precisely blocked by specific antagonists.
Examples:
- Acetylcholine receptors can be blocked.
- Adrenaline receptors can be blocked.
- Histamine acts on specific H1, H2, H3, and H4 receptors (allergy medicines block H1).
- Dopamine acts on D1–D5 receptors (antipsychotic drugs block these).
- Morphine acts on specific opioid receptors named μ (mu), κ (kappa), and δ (delta).
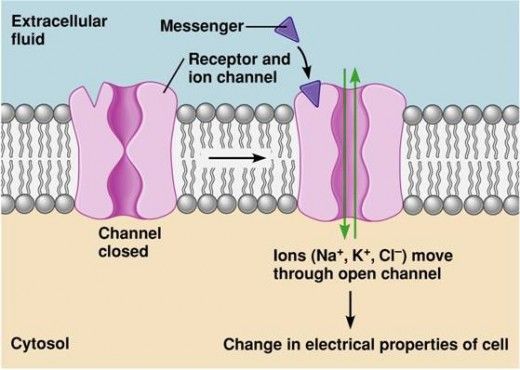
Target II: Ion Channels
Cells use electrical charges to communicate, especially nerves and muscles. They do this by moving ions (like Sodium, Calcium, Potassium, and Chloride) in and out of the cell through specialized protein gates called Ion Channels.
There are two main types of ion channels:
- Ligand-gated (ionotropic) channels: These are locked gates that only open when a specific chemical key (an agonist) binds directly to the receptor on the gate.
- Voltage-gated channels: These gates do not need a chemical key. Instead, they sense the electrical charge of the cell. They open or close in response to changes in the membrane potential (electrical voltage).
How Drugs Act on Ion Channels:
- Direct Action: The drug physically binds directly to the channel protein itself, acting like a plug to block it, or locking it in an open position.
- Indirect Action: The drug binds to a separate receptor nearby, which then uses a messenger (like a G-protein) to tell the ion channel to open or close.
Examples of Drugs Acting on Ion Channels:
- Voltage-gated sodium channels: These are blocked by local anesthetics (e.g., lidocaine). By blocking sodium from entering the nerve, the nerve cannot send a pain signal to the brain.
- L-type calcium channels: These are inhibited by dihydropyridines (a class of vasodilators, e.g., nifedipine). By blocking calcium from entering blood vessel muscles, the vessels relax, heavily lowering blood pressure.
- GABA receptor–chloride channel system: This is modulated by benzodiazepines (tranquillizers, e.g., diazepam). Diazepam binds to the channel, helping it open wider to let negatively charged chloride ions into the brain cell, severely calming and slowing down brain activity.
- ATP-sensitive potassium channels (KATP): Located in the pancreatic β-cells. These are blocked by sulfonylureas (diabetes medications). Blocking potassium from leaving the cell forces the pancreas to release stored insulin into the blood.
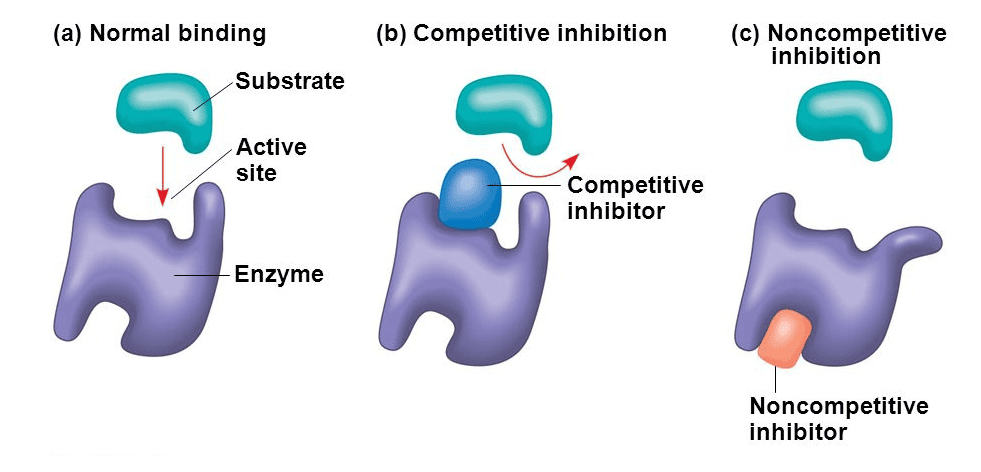
Target III: Enzymes
Enzymes are biological catalysts that speed up chemical reactions in the body (building things up or breaking them down). Many drugs act specifically as enzyme inhibitors.
- I. Competitive, Reversible Inhibition: The drug temporarily fights the natural substance for the active spot on the enzyme. If the drug wins, the enzyme halts. Because it is reversible, the effect wears off as the drug leaves the body.
- Example: Neostigmine. It reversibly inhibits the enzyme acetylcholinesterase (the enzyme that destroys acetylcholine). This allows acetylcholine to build up and help patients with severe muscle weakness.
- II. Irreversible, Non-Competitive Inhibition: The drug permanently binds to the enzyme, destroying its ability to function forever. The body must physically build entirely new enzymes to recover.
- Example: Aspirin. It permanently inhibits the cyclo-oxygenase (COX) enzyme, permanently stopping the production of chemicals that cause pain and inflammation.
- III. False Substrates: The drug tricks the enzyme. The enzyme thinks the drug is a normal building block and tries to process it, producing abnormal, broken metabolites that disrupt cell pathways.
- Example: Fluorouracil. This is an anticancer drug. Cancer cells take it up thinking it is a building block for DNA, but it ruins their DNA production, killing the cancer cell.
Target IV: Carrier Molecules (Transporters)
Many essential molecules in the body are polar ions or small organic molecules. Because they are polar, they cannot diffuse freely through the fatty cell membrane. They require special "taxi cabs" called carrier molecules or transporters to carry them across the membrane.
Natural examples of these transporters include Glucose and amino acid transporters, Ion transporters, and Neurotransmitter transporters (which vacuum up used neurotransmitters like choline, noradrenaline, serotonin, and glutamate from the brain synapses to be recycled).
Amine Transporters (Distinct from Receptors)
These belong to a separate structural family from receptors. Carrier proteins have highly specific recognition sites for their substrates. Drugs can target these exact sites to block transport.
Crucial Examples of Drugs Targeting Carriers/Transporters:
Block the reuptake transporters for noradrenaline and serotonin, leaving more of these mood-boosting chemicals in the brain.
Blocks the reuptake transporters for dopamine, noradrenaline, and serotonin, causing a massive, temporary high.
(e.g., Fluoxetine). Specifically inhibit the serotonin transporter (SERT), leading to increased serotonin at synapses to treat depression.
Inhibits the H⁺/K⁺-ATPase proton pump (a specific transporter) in the stomach's parietal cells, stopping severe acid reflux.
(e.g., digoxin): Inhibit the Na⁺/K⁺-ATPase pump in heart cells. This indirect action forces intracellular calcium levels to rise, causing the heart to pump with much greater force.
(e.g., Furosemide): Inhibit the Na⁺-K⁺-2Cl⁻ cotransporter (NKCC2) in the renal tubules of the kidneys, causing massive water loss (urine).
(e.g., Hydrochlorothiazide): Block the Na⁺-Cl⁻ cotransporter (NCC) in the distal tubule of the kidney.
(e.g., Dapagliflozin): Block the sodium-glucose cotransporter-2 in the kidney. This prevents the kidney from reabsorbing sugar, leading to increased glucose excretion in the urine to treat diabetes.
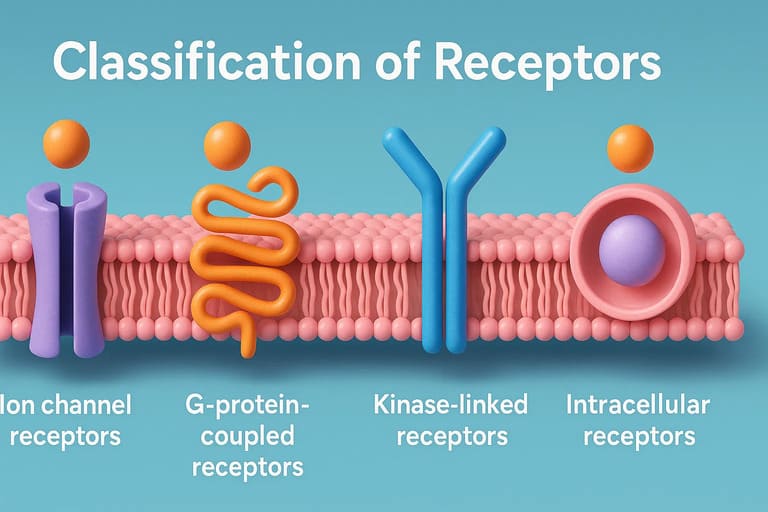
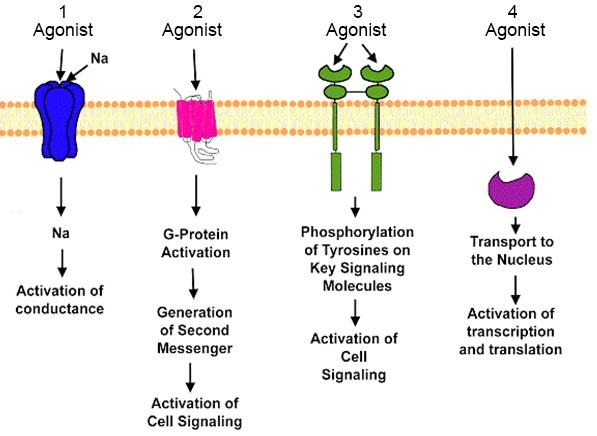
The Four Main Types of Receptors
Receptors are not all built the same. They differ heavily in their physical structure, their internal signaling mechanism, and their speed of response. They are divided into four main classes:
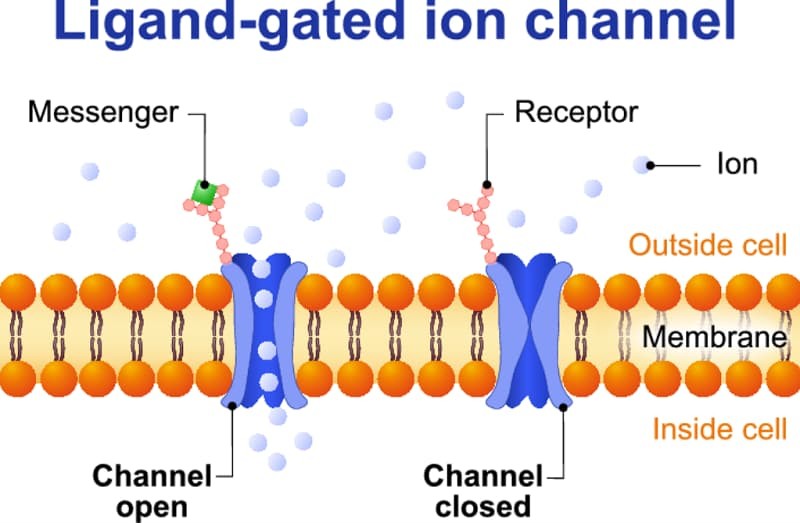
Type 1: Ligand-Gated Ion Channels (Ionotropic Receptors)
- Structure: Membrane proteins containing an extracellular ligand-binding site physically attached to an ion channel pore.
- Function: Mediate incredibly fast synaptic transmission between nerves.
- Operating time: Milliseconds (the fastest receptor type).
- Examples:
- The Nicotinic Acetylcholine Receptor (nAChR): Composed of exactly five protein subunits forming a ring structure. These subunits are of four different types: α (alpha), β (beta), γ (gamma), and δ (delta). Each subunit is embedded in the cell membrane, forming a central pore. Mechanism: The receptor has exactly two binding sites for acetylcholine (ACh). The channel opens only when both binding sites are occupied by ACh molecules.
- GABAA receptor
- Glutamate (NMDA) receptor
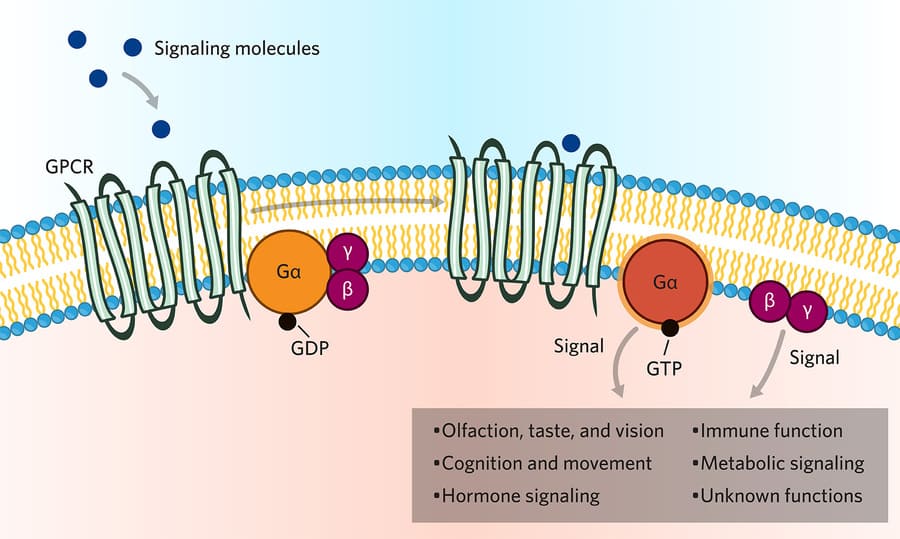
Type 2: G-Protein–Coupled Receptors (GPCRs) / Metabotropic Receptors
- Structure: A single polypeptide chain winding through the cell membrane exactly seven times (seven transmembrane domains). A large loop on the inside of the cell interacts with a G-protein.
- Function: These are membrane receptors linked to intracellular effector systems (enzymes inside the cell) through intermediary G-proteins. They form the largest receptor family in the human body.
- Operating time: Seconds.
- Effectors: They mediate the actions of many hormones, peptides, catecholamines, and slow neurotransmitters.
- Examples: Muscarinic acetylcholine receptors, adrenoceptors, and chemokine receptors. While multiple subtypes exist, they all share the exact same basic structural 7-transmembrane framework.
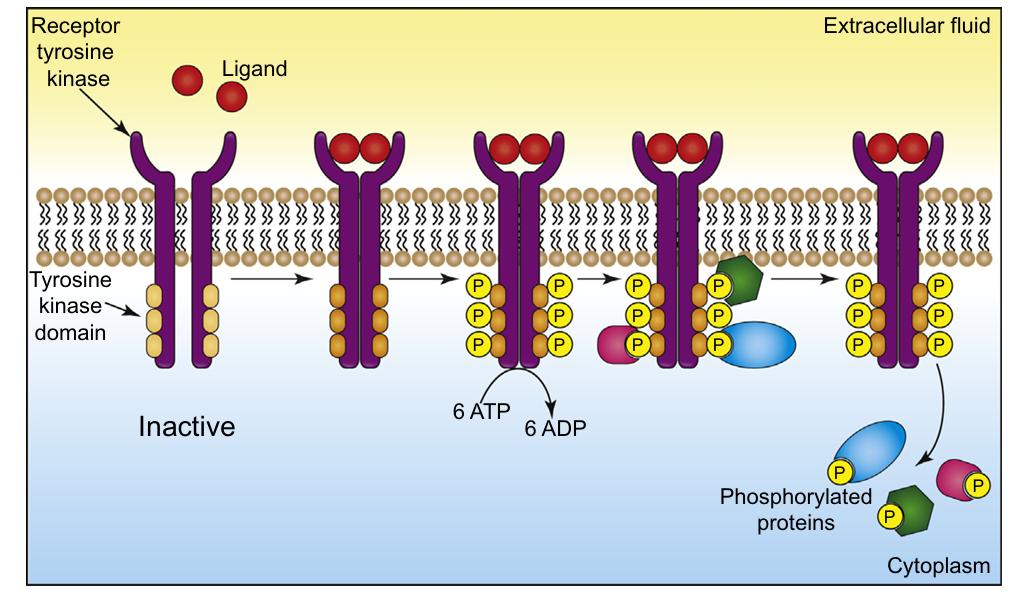
Type 3: Kinase-Linked and Related Receptors
- Structure: A large and extremely heterogeneous group. They consist of an extracellular ligand-binding domain, a single transmembrane helix, and an intracellular domain that acts directly as an enzyme.
- Function: The inside of the receptor has enzymatic activity (e.g., protein kinase or guanylyl cyclase activity) or is tightly coupled to intracellular enzymes.
- Operating time: Hours.
- Effectors: They mainly respond to protein mediators like peptide hormones and growth factors.
- Subclasses and Examples:
- Receptor Tyrosine Kinases (RTKs): e.g., the Insulin receptor, Epidermal Growth Factor (EGF) receptor.
- Receptor Serine/Threonine Kinases: e.g., receptors for Transforming Growth Factor-β (TGF-β).
- Cytokine Receptors: e.g., receptors for interleukins, growth hormone, erythropoietin. These are strongly associated with Janus kinases (JAKs).
- Receptor Guanylyl Cyclases: e.g., atrial natriuretic peptide (ANP) receptor.
Type 4: Nuclear (Intracellular) Receptors
- Structure & Location: Unlike the other three, these are not stuck in the cell membrane. Although termed "nuclear" receptors, some float freely in the cytosol (the cell fluid) and only translocate (move) into the nucleus after the ligand binds to them.
- Function: They strictly regulate gene transcription. They act as transcription factors, physically binding to specific DNA sequences to turn gene expression on or off.
- Operating time: Hours to days (This is a very long-term process because building new proteins from DNA takes significant time).
- Examples: Receptors for steroid hormones (glucocorticoids, mineralocorticoids, androgens, estrogens), thyroid hormones, retinoic acid, and vitamin D.

The Receptor Concept and Drug Interactions
History of the Receptor Concept
- The initial idea of drugs acting on invisible specific targets (receptors) is credited to John Langley (1878) while he was studying the antagonism between two plant chemicals, atropine and pilocarpine, and how they induced or blocked salivation.
- The actual term receptor was introduced in 1909 by Paul Ehrlich. He proposed a famous rule: drugs exert therapeutic effects only if they possess the "right sort of affinity."
- Ehrlich defined a receptor as: “that combining group of the protoplasmic molecule to which the introduced group is anchored.”
- Today, we know that from a numerical standpoint, proteins are the most important class of drug receptors. This encompasses hormone receptors, growth factor receptors, transcription factors, neurotransmitter receptors, and cellular enzymes.
Drug–Receptor Interaction and Bonding
When a drug finds its receptor, it must stick to it. Drug binding to receptors can involve various chemical forces:
- Noncovalent bonds: Ionic bonds, hydrogen bonding, hydrophobic interactions, and van der Waals forces. Most drug–receptor interactions involve multiple types of these weak, temporary bonds.
- Covalent bonds: These are incredibly strong, permanent chemical bonds. Covalent binding usually results in prolonged, irreversible drug action (like Aspirin permanently breaking COX enzymes). *Note: Extremely high-affinity noncovalent interactions can sometimes behave as if they are irreversible because they hold on so tightly.*
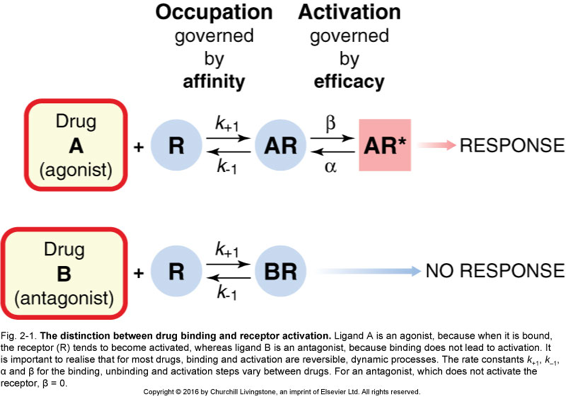
Affinity vs. Intrinsic Activity (The Key and Lock Analogy)
Drug-receptor interaction occurs in two distinct steps:
- Binding (Affinity): This is the ability of the drug to locate, physically bind to, and maintain a connection with the receptor. It is determined by the chemical shape and forces. Analogy: Affinity is how well the key fits into the lock.
- Generation of a Response (Intrinsic Activity): This reflects the ability of the drug-receptor structure to actually produce a biological pharmacological effect once connected. Analogy: Intrinsic activity is whether or not the key can actually turn and open the door.
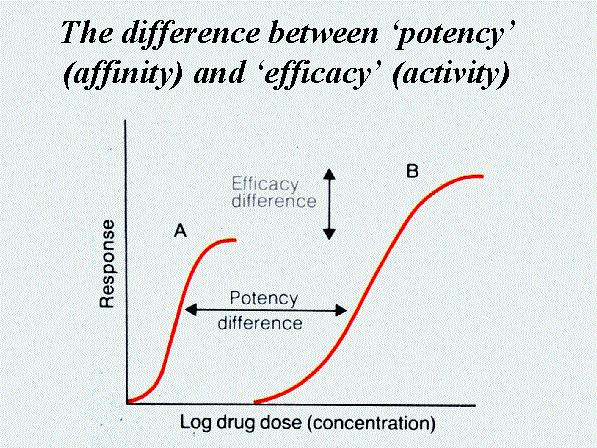
Potency and Efficacy
These two terms are strictly different and frequently tested:
- Potency: Refers to the amount (dose/weight) of drug strictly required to produce a given effect. A drug is considered highly potent if it produces a significant response at a very low dose (e.g., 1 milligram). Potency is important for doctors when determining the appropriate dosage to prescribe.
- Efficacy: Refers to the maximum or peak response a drug can physically produce, regardless of how high you push the dose. It is a critical factor in drug selection. If a patient is in severe pain, you need a drug with high efficacy (like morphine), not just a highly potent drug that has a low ceiling of effect.
Theories Explaining the Intensity of Drug Response
When a drug binds, how does the body decide how intense the reaction should be? Three main theories attempt to explain this mathematical relationship:
- Rate Theory: The response depends solely on the speed (rate) at which the drug associates with and dissociates from the receptor, rather than how many receptors are occupied at a given time.
- Drug activity depends on k₁ (the rate of association/binding) and k₂ (the rate of dissociation/breaking apart).
- Agonists have high association and high dissociation rates, leading to a rapid turnover of binding events, which creates a strong response.
- Drug-Induced Protein Change Theories: The drug induces a physical conformational (shape) change in the receptor protein upon binding. This physical structural alteration initiates the biological response.
- Agonists cause temporary structural changes that alter cell membrane permeability to produce a response. Antagonists cause changes that block further binding.
- Receptor Occupation Theory: The simplest theory. The response is strictly, directly proportional to the physical number of receptors occupied by the drug. A maximal effect occurs when 100% of all available receptors are occupied.
Receptors are central to pharmacology. They mediate most drug actions, determine the selectivity of both therapeutic and toxic effects, and dictate the exact mathematical quantitative relationship between the drug dose and the pharmacologic response.
Types of Drugs Based on Receptor Interaction
A drug, hormone, or neurotransmitter that binds to its specific receptor, successfully activates it, and initiates a full response. It has both high affinity and high intrinsic activity.
Examples: Acetylcholine, noradrenaline.
A drug that binds firmly to a receptor but does not activate it. Instead, it acts as a shield, preventing the action of a natural agonist by blocking receptor access. A pure competitive antagonist has high affinity but absolutely zero intrinsic activity of its own.
Examples: Atropine, Naloxone.
A drug that binds to a receptor and activates it, but mathematically produces a weaker (submaximal) response compared to a full agonist. Its intrinsic activity is greater than 0 but less than 1.
Unique feature: Under certain conditions, if a full agonist is already present, adding a partial agonist will actually act as an antagonist because it steals the receptor seat from the full agonist, resulting in a lower overall response.
Example: Aripiprazole (an atypical antipsychotic). It acts as a partial agonist at dopamine receptors—it inhibits severely overactive dopaminergic pathways (calming the brain) while gently stimulating underactive ones.
A drug that produces an effect physically opposite to that of an agonist.
Crucial Clarification: An inverse agonist does not simply produce an effect "opposite to the agonist" in a general behavioral sense. Instead, it produces an effect opposite to the receptor's constitutive (basal/resting) activity. While a normal antagonist just sits there and blocks, an inverse agonist actively shuts down the receptor's baseline hum.
Example: Benzodiazepines (agonists) on GABA receptors cause severe sedation and anxiolysis. Inverse agonists (like β-carbolines) bind to the exact same receptor but actively cause extreme stimulation, anxiety, and convulsions.
A drug that acts as a full agonist at one specific receptor subtype, but simultaneously acts as an antagonist at a different related subtype.
Example: Some opioids, such as pentazocine and nalorphine. They can produce bizarre psychotomimetic (hallucinatory) effects that are uniquely not reversed by naloxone, and they may instantly precipitate withdrawal symptoms in opioid-dependent patients, heavily limiting their clinical use.
Mechanism of Signal Transduction: GPCRs in Detail
Let us take a deeper look at the Type 2 receptors: G-protein–coupled receptors (GPCRs), also known as metabotropic receptors.
GPCRs regulate cellular functions by activating intracellular signaling pathways. The physical connection between the receptor on the outside of the cell and the signaling enzymes on the inside of the cell is mediated by a middle-man known as a G-protein.
Facts about G-Proteins:
- They act as physical intermediaries between the receptor and the effector targets (enzymes or ion channels).
- They are named "G-proteins" because of their interaction with guanine nucleotides (GTP and GDP).
- They are heterotrimeric proteins, meaning they are built from three different subunits named α (alpha), β (beta), and γ (gamma).
- The α-subunit possesses GTPase activity, which acts as a timer to regulate and eventually shut off the signaling.
The Three Main Classes of G-Proteins
Depending on which specific G-protein the receptor is attached to, the cell will do entirely different things:
- Gs (Stimulatory): Stimulates the enzyme Adenylyl Cyclase → Increases cAMP levels in the cell.
- Gi (Inhibitory): Inhibits the enzyme Adenylyl Cyclase → Decreases cAMP levels in the cell.
- Gq: Activates the enzyme Phospholipase C (PLC) → Increases IP₃ and DAG → Increases Calcium (Ca²⁺) levels.
- (Minor class) G12/13: Activates RhoGEFs (RhoA) to regulate the cellular cytoskeleton, cell shape, and migration. It does not use classical second messengers.
Target 1: The Adenylyl Cyclase / cAMP System (Gs and Gi)
Many drugs regulate the activity of the membrane-bound enzyme adenylyl cyclase. Here is the step-by-step pathway:
- A drug binds to a Gs-coupled receptor.
- The Gs protein is activated and turns on the enzyme Adenylyl Cyclase.
- Adenylyl Cyclase rapidly converts regular ATP energy molecules into a second messenger called cAMP.
- Rising cAMP levels activate Protein Kinase A (PKA).
- PKA goes on to phosphorylate proteins, increasing heart rate, increasing lipolysis (fat breakdown), and changing gene expression.
To stop the signal: cAMP is continuously degraded and destroyed by maintenance enzymes called phosphodiesterases (PDEs), which turn it into inactive 5′-AMP.
Conversely, if a drug binds to a Gi-coupled receptor, the exact opposite happens. Adenylyl cyclase is blocked, cAMP drops, PKA activity drops, and protein phosphorylation decreases.
Target 2: The Phospholipase C / IP₃–DAG System (Gq)
If a drug binds to a Gq-coupled receptor, a completely different signaling pathway occurs:
- A drug binds to a Gq-coupled receptor.
- The Gq protein activates a different membrane enzyme called Phospholipase C (PLC-β).
- PLC acts like a pair of scissors. It cuts a fat molecule in the membrane called PIP₂ into two distinct second messengers: IP₃ and DAG.
- IP₃ (Inositol trisphosphate): Diffuses deep into the cytoplasm and triggers massive Calcium (Ca²⁺) release from the cell's storage unit (the endoplasmic reticulum).
- DAG (Diacylglycerol): Remains stuck in the cell membrane and activates Protein Kinase C (PKC).
The combined action of skyrocketing Calcium and PKC activation causes intense cellular effects, primarily smooth muscle contraction and glandular secretion.
Target 3: Direct Ion Channel Regulation by GPCRs
Sometimes GPCRs do not use complex enzymes. Certain GPCRs directly regulate ion channels using the G-protein's leftover βγ (beta-gamma) subunits.
- Potassium (K⁺) channels: These are often physically opened by Gi-coupled receptors. This causes potassium to flood out, leading to hyperpolarization and deeply lowered excitability. (Example: Muscarinic M₂ receptors doing this in the heart causes the heart rate to slow down).
- Calcium (Ca²⁺) channels: These are often physically inhibited (closed) by Gi-coupled receptors. This reduces calcium entry, which instantly stops the nerve from releasing neurotransmitters. (Example: Presynaptic autoreceptors shutting down the nerve terminal).
Through these intricate systems, GPCRs seamlessly control membrane potential, neuronal firing, massive muscle contractions, and cellular secretion.