Introduction to Hemostasis
Hemostasis is the physiological process that stops bleeding at the site of vascular injury while maintaining normal blood flow elsewhere. It involves interactions between blood vessels, platelets, and coagulation factors. Dysregulation leads to hemorrhage (excessive bleeding) or thrombosis (inappropriate clotting).
Platelets (thrombocytes) are small, anucleated cell fragments that play a central role in primary hemostasis – the initial formation of a platelet plug at the site of injury.
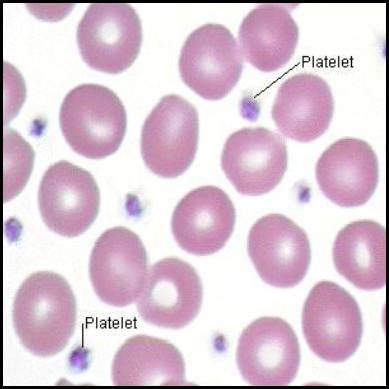
I. Morphology of Platelets
Physical Traits
- Size/Shape: Tiny (2-4 µm), discoid (lens-shaped) when inactive. Upon activation, they become spherical with pseudopods (finger-like projections) to enhance adhesion.
- Anucleated: Lack a nucleus; cannot synthesize proteins. Limited lifespan (7-10 days).
Membrane
Rich in glycoproteins (e.g., GP Ib/IX/V, GP Ia/IIa, GP IIb/IIIa) acting as receptors for adhesion molecules (vWF, collagen, fibrinogen).
Cytoplasmic Granules
The cytoplasm contains critical granules and organelle systems.
Alpha-granules
Contain proteins for adhesion/coagulation:
- Fibrinogen
- von Willebrand factor (vWF)
- Platelet factor 4 (PF4)
- PDGF, P-selectin
Dense (delta) granules
Contain non-protein activators:
- ADP, ATP
- Serotonin
- Calcium
Lysosomes
Contain hydrolytic enzymes for digesting material.
II. Formation of Platelets (Thrombopoiesis)
Occurs in bone marrow, regulated by Thrombopoietin (TPO).
Hematopoietic Stem Cells (HSCs) → Common Myeloid Progenitor (CMP).
Progenitor undergoes endoreduplication (DNA replication without division), becoming polyploid.
Largest marrow cell (up to 100 µm). Highly lobulated nucleus.
Megakaryocytes extend proplatelets into sinusoidal capillaries. Blood shear flow fragments these into thousands of platelets (1,000-3,000 per megakaryocyte).
III. Function of Platelets in Hemostasis (Primary Hemostasis)
1. Adhesion
- Injury exposes subendothelial collagen.
- Platelets adhere via GP Ib receptor binding to von Willebrand factor (vWF) (bridge between platelet and collagen).
- Direct binding via GP Ia/IIa also occurs.
- Result: Anchors platelets to injury site.
2. Activation
Triggered by adhesion, Thrombin, and ADP. Causes shape change (discoid → spherical + pseudopods) and granule release.
Key Molecules Released:- ADP: Potent activator, recruits more platelets.
- Thromboxane A2 (TxA2): Synthesized via COX-1; powerful vasoconstrictor and aggregator.
- Serotonin: Vasoconstriction.
- vWF/Fibrinogen: Aid further adhesion/aggregation.
3. Aggregation
- Activated platelets express GP IIb/IIIa receptor.
- Fibrinogen acts as a bridge, binding to GP IIb/IIIa on adjacent platelets.
- Links platelets together to form the primary hemostatic plug.
4. Procoagulant Activity
Activated platelets provide a negatively charged phospholipid surface (phosphatidylserine). This surface concentrates coagulation factors (Tenase/Prothrombinase complexes), accelerating Thrombin generation to convert fibrinogen to fibrin, stabilizing the plug.
Summary of Platelet Function
When a vessel is damaged, platelets:
- Adhere to exposed matrix.
- Activate (shape change + release substances).
- Aggregate to form primary plug.
- Provide surface for Secondary Hemostasis (Coagulation Cascade).
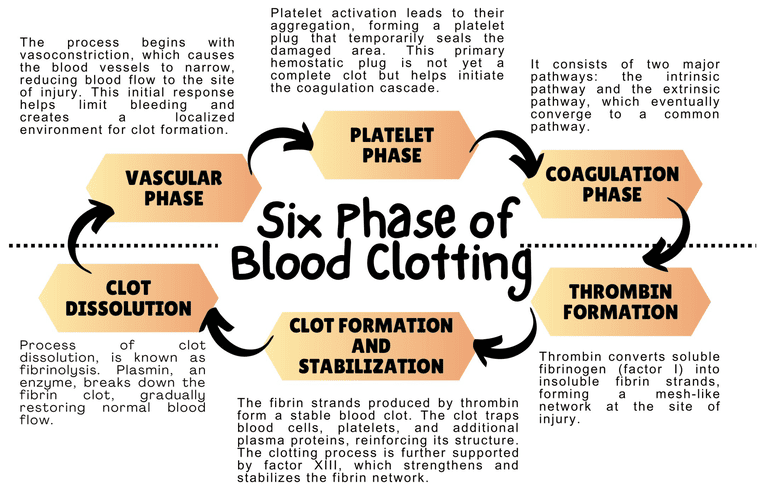
Steps and Components of the Coagulation Cascade
While primary hemostasis (platelet plug formation) provides an initial, temporary seal at the site of vascular injury, it is not strong enough to withstand arterial pressure or provide long-term protection. Secondary hemostasis reinforces the platelet plug with a meshwork of fibrin, a strong, insoluble protein. This process is known as blood coagulation or the coagulation cascade, and it involves a series of enzymatic reactions involving plasma proteins called coagulation factors.
I. Overview
The coagulation cascade is traditionally described as having two main pathways that converge on a common pathway. However, a more modern and physiologically relevant view is the cell-based model. We will present both models for a comprehensive understanding.
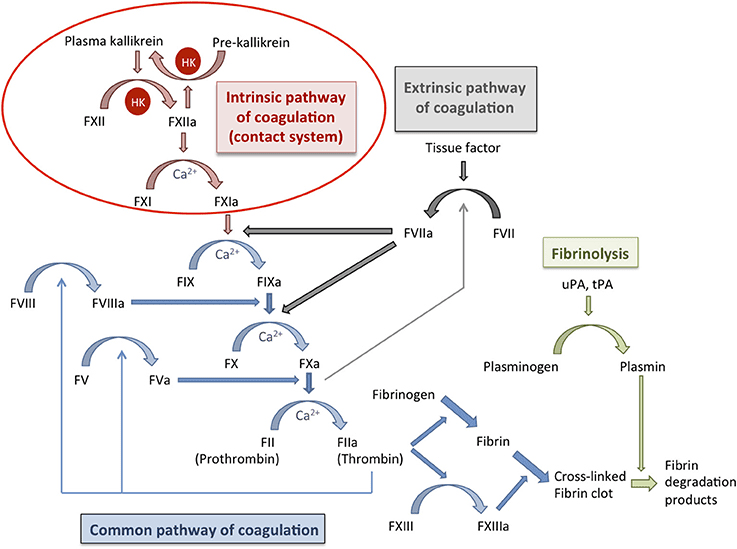
II. Traditional Model: Intrinsic, Extrinsic, and Common Pathways
This model helps to understand individual factors and their interactions, especially in laboratory testing.
1. Extrinsic Pathway (Initiation)
Initiated when blood is exposed to Tissue Factor (TF), expressed by subendothelial cells (fibroblasts, smooth muscle) upon injury.
Step 1: TF binds to circulating Factor VII (VIIa) → Forms TF-VIIa complex.
Step 2: TF-VIIa complex activates Factor X to Xa and Factor IX to IXa.
Rapid pathway; primarily responsible for initiation.
2. Intrinsic Pathway (Amplification)
Activated by contact of Factor XII with negatively charged surfaces (collagen, platelets) or by XIIa itself.
Step 1: Factor XII → XIIa.
Step 2: XIIa activates Factor XI → XIa.
Step 3: XIa activates Factor IX → IXa.
Step 4: IXa + Factor VIIIa (activated by thrombin) → Tenase Complex (IXa/VIIIa).
The Tenase complex activates Factor X → Xa.
Slower pathway; significant contribution to amplification.
3. Common Pathway
Both pathways converge at the activation of Factor X.
Assembles on activated platelet surfaces.
Central event of the cascade.
- Converts Fibrinogen (I) → Fibrin monomers.
- Activates Factor XIII → XIIIa.
- Activates cofactors V and VIII.
- Activates Factor XI (feedback amplification).
- Activates platelets (positive feedback).
III. Cell-Based Model of Coagulation
This model emphasizes the role of cellular surfaces (TF-bearing cells and activated platelets) and occurs in three overlapping phases.
1. Initiation Phase (on TF-bearing cells)
- Vascular injury exposes TF on subendothelial cells.
- TF binds VIIa → TF-VIIa.
- TF-VIIa activates small amounts of X to Xa and IX to IXa.
- Xa + Va generates a small "Thrombin Spurt". Crucial for activating platelets/cofactors.
2. Amplification Phase (on Activated Platelets)
- The initial thrombin activates platelets (shape change, exposure of phosphatidylserine).
- Thrombin activates cofactors V, VIII, and Factor XI.
- Activated platelets provide negatively charged phospholipid surface.
- Factor IXa binds VIIIa on platelet surface → Tenase Complex.
- Tenase efficiently converts large amounts of X → Xa.
3. Propagation Phase (on Activated Platelets)
- Large amounts of Xa + Va assemble on platelet surface → Prothrombinase Complex.
- Converts massive amounts of prothrombin → thrombin. Result: "Thrombin Burst".
- Thrombin burst rapidly converts fibrinogen to fibrin, activates XIIIa (cross-linking), and further activates platelets → Robust, stable clot.
IV. Key Coagulation Factors & Components
Plasma proteins, mostly synthesized in the liver.
Factors II, VII, IX, X, Protein C, Protein S. Require Vitamin K for synthesis in liver.
Factors I (Fibrinogen), V, VIII, XIII. Consumed during coagulation.
Factors XII, XI, PK, HMWK. Intrinsic pathway initiation.
Essential cofactors for activation/function of several factors, particularly for assembly of Tenase/Prothrombinase complexes on phospholipid surfaces.
Generate a stable, cross-linked fibrin mesh that traps RBCs/cellular elements, providing mechanical strength to the platelet plug and forming a definitive blood clot.
Regulation of Clotting and Fibrinolysis
Hemostasis is a delicate balance. While rapid clot formation is vital to stop bleeding, uncontrolled or excessive clotting can lead to thrombosis, blocking blood vessels and causing severe damage (e.g., heart attack, stroke). Therefore, the body has sophisticated mechanisms to regulate the coagulation cascade and dissolve clots once they are no longer needed.
I. Regulation of Coagulation (Anticoagulation Systems)
These systems work to limit the size and propagation of the clot to the site of injury, preventing it from spreading unnecessarily.
1. Antithrombin (AT)
Mechanism: Major plasma protein that inactivates several factors, particularly Thrombin (IIa), Xa, and lesser amounts of IXa, XIa, and XIIa.
Action: Forms a stable, irreversible complex with these serine proteases, rendering them inactive.
2. Protein C System
Components: Thrombomodulin, Protein C, and Protein S.
Activation:- Thrombin binds to Thrombomodulin (receptor on healthy endothelium).
- This complex activates Protein C into Activated Protein C (APC).
- APC + cofactor Protein S inactivates cofactors Va and VIIIa (via cleavage).
- Shuts down prothrombinase and tenase complexes, stopping thrombin generation.
Clinical Relevance: Deficiency in Protein C or S increases thrombosis risk.
3. Tissue Factor Pathway Inhibitor (TFPI)
Mechanism: Directly inhibits the initial step of the extrinsic pathway.
Action: Binds and inactivates Factor Xa. The TFPI-Xa complex then binds and inactivates the TF-VIIa complex.
Result: "Turns off" the tissue factor pathway, limiting the initial thrombin burst.
Flowing blood dilutes activated factors, washing them away from the injury site preventing expansion.
Liver clears activated factors and inhibitors from circulation to maintain balance.
II. Clot Dissolution (Fibrinolysis)
Once repair occurs, the stable fibrin clot must be removed (fibrinolysis) to restore flow.
Key Enzyme: Plasmin
- Mechanism: Serine protease that cleaves fibrin and fibrinogen, breaking the meshwork.
- Formation: Circulates as inactive Plasminogen.
- Activation: Converted to Plasmin by Plasminogen Activators.
Plasminogen Activators
- Released from damaged endothelium.
- High affinity for fibrin.
- Binds fibrin within clot → activates plasminogen locally.
- Found in tissues/fluids (urine).
- Converts plasminogen to plasmin.
- Role in local fibrinolysis and tissue remodeling.
Inhibitors of Fibrinolysis
Ensures clot doesn't dissolve prematurely.
- Plasminogen Activator Inhibitor-1 (PAI-1): Inhibits t-PA and u-PA (reduces plasmin generation).
- Alpha-2-antiplasmin (α2AP): Primary inhibitor of free plasmin in circulation. Prevents systemic fibrinogen breakdown.
- TAFI (Thrombin Activatable Fibrinolysis Inhibitor): Activated by thrombin. Removes lysine residues from fibrin, making it resistant to plasmin. Links coagulation to fibrinolysis regulation.
Products of Fibrinolysis
Breakdown produces soluble Fibrin Degradation Products (FDPs).
D-Dimer: Specific FDP formed when cross-linked fibrin (by XIIIa) is degraded.
Clinical Significance: Elevated levels indicate recent/ongoing clot formation and breakdown.
Summary of Regulation and Fibrinolysis
- Anticoagulation systems (AT, Protein C, TFPI) prevent expansion beyond injury.
- Fibrinolysis (Plasminogen/Plasmin via t-PA/u-PA) ensures timely removal.
- The balance is crucial for vascular patency and preventing bleeding/thrombosis.
Laboratory Tests and Disorders of Hemostasis
Laboratory tests distinguish between bleeding and clotting disorders, identify specific deficiencies, and guide therapy. They are generally categorized by the phase of hemostasis they assess.
I. Tests of Primary Hemostasis (Platelet Function)
Evaluate platelet number, adhesion, and aggregation.
1. Platelet Count
Normal: 150k - 450k/µLBleeding (petechiae, purpura). Causes: Marrow failure, ITP/TTP, splenomegaly.
Risk of thrombosis or paradoxical bleeding (dysfunction).
Screening test simulating vessel injury. Detects vWD, aspirin use, intrinsic defects.
Definitive test. Measures response to agonists (ADP, collagen, ristocetin). Diagnoses vWD, Bernard-Soulier.
Note: Bleeding Time is largely historical and replaced by PFA-100.
II. Tests of Secondary Hemostasis (Coagulation Cascade)
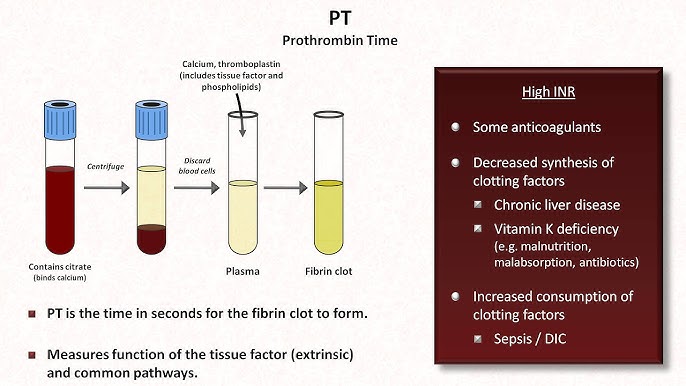
Prothrombin Time (PT) & INR
Extrinsic/CommonNormal PT: 10-14 sec. | Normal INR: 0.8-1.2
Prolonged in: Deficiency of VII, X, V, II, Fibrinogen. Liver disease, Vit K deficiency, Warfarin therapy.
INR Use: Standardizes PT for monitoring Warfarin (Target usually 2.0-3.0).
Activated Partial Thromboplastin Time (aPTT)
Intrinsic/CommonNormal Range: 25-35 sec.
Prolonged in: Deficiency of XII, XI, IX, VIII, X, V, II. Heparin therapy, Hemophilia A/B, Lupus anticoagulant.
Use: Monitoring Heparin therapy.
Measures Fibrinogen → Fibrin conversion. Prolonged by low fibrinogen, heparin, FDPs.
Normal: 200-400 mg/dL. Low in DIC/Liver disease. High in inflammation.
III. Tests of Fibrinolysis (D-Dimer)
D-Dimer
Specific degradation product of cross-linked fibrin.
- Elevated: Indicates clot formation/breakdown. Screening for DIC, PE, DVT.
- Negative: High negative predictive value to rule out DVT/PE in low-risk patients.
Common Disorders of Hemostasis
I. Primary Hemostasis Disorders (Platelet/Vessel)
Symptoms: Mucocutaneous bleeding (petechiae, epistaxis).
- Decreased Production: Marrow suppression, leukemia, B12/folate deficiency.
- Increased Destruction: ITP (Autoimmune), TTP/HUS (Microangiopathic).
- Sequestration: Splenomegaly.
- Inherited: Glanzmann's (GP IIb/IIIa), Bernard-Soulier (GP Ib).
- Acquired: Aspirin/NSAIDs, Uremia.
Most common inherited bleeding disorder. Deficiency/defect in vWF (platelet adhesion + Factor VIII carrier).
II. Secondary Hemostasis Disorders (Coagulation Factors)
Symptoms: Deep tissue bleeding (hemarthroses, hematomas).
X-linked recessive. Deep bleeding.
Labs: Prolonged aPTT, Normal PT.
Affects II, VII, IX, X. Diet/Malabsorption/Warfarin.
Labs: Prolonged PT (sensitive) & aPTT.
Reduced synthesis of factors. Bleeding + Thrombosis risk.
Labs: Prolonged PT/aPTT, Low Platelets.
Widespread activation (sepsis/trauma) → Consumption of factors → Bleeding + Clotting.
Labs: ↓ Platelets, ↑ PT/aPTT, ↓ Fibrinogen, ↑ D-Dimer.
III. Thrombotic Disorders (Thrombophilia)
- Factor V Leiden: Resistance to APC (Most common).
- Prothrombin Gene Mutation: High prothrombin.
- Deficiencies: Antithrombin, Protein C, Protein S.
- Antiphospholipid Syndrome (APS): Autoimmune. Paradoxical prolonged aPTT.
- Others: Malignancy, Pregnancy, Immobilization, HIT (Heparin-Induced).
Platelets and Hemostasis
Test your knowledge with these 25 questions.
Platelets and Hemostasis Quiz
Question 1/25
Quiz Complete!
Here are your results, .
Your Score
23/25
92%